用于提高突变评估准确性的方法和装置与流程
相关申请的交叉引用本申请要求2015年2月26日提交的美国临时专利申请第62/120923号的优先权权益,其整体通过引用并入本文。
背景技术:
:a.
技术领域:
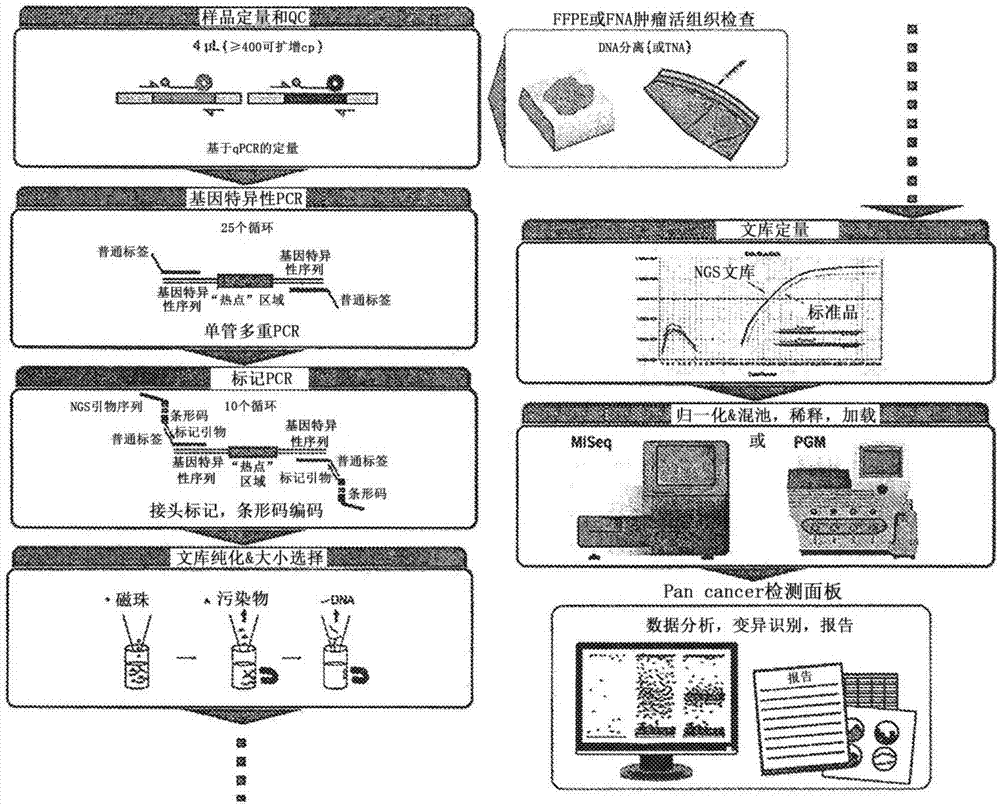
本发明一般地涉及核酸分析领域,更具体地涉及将可行模板计数参数并入基于计算机的变异识别模型,其可以与涉及核酸分子的化学和/或物理操作的分析结合使用。实施方案包括涉及利用可行模板计数评估以提高变异识别准确性的变异识别算法的方法和产品。b.相关技术说明许多临床样本可用性的限制推动了对低dna输入到分子分析中的需要。例如,下一代测序(ngs)是尖端技术,其可以推进深度分子图谱所需的输入dna材料的界限,特别是在癌症中(beltran等人,2013,menon等人,tuononen等人,2013,hadd等人,2013)。ngs具有精确地检测点突变、结构变异、拷贝数变化、甲基化状态和基因表达的能力,是多层面的且通用的工具;然而,在肿瘤样品的ngs中的高灵敏度、高特异性单核苷酸变异(snv)识别是具有挑战性的问题。输入样品通常是异质的,其含有正常材料和肿瘤材料的混合物,其中所述肿瘤材料本身可以由细胞的异质群体组成。因此,任何变异检测算法实现高灵敏度并具有很低变异频率以避免漏掉真正的突变是至关重要的。变异识别进一步受到将背景噪声提高至生物变异的同等水平的低质和低量输入的挑战。因此,用于snv识别的任何方法还必须实现高特异性,以避免过度识别样品。特别挑战类型的输入样品包括福尔马林固定石蜡包埋(ffpe)的肿瘤dna。ffpe表现出对突变检测的双重挑战,即对于抵抗pcr扩增的低模板输入量连同来自固定和包埋处理的模板损伤的要求。此外,低质量ffpedna可以触发等位基因丢失和产生不准确的结果(didelot等人,2013,akbari等人,2005)。为了着手解决一些建立可以指导可靠测序结果的质量控制指标的挑战,实体如临床测试下一代测序标准(nex-stoct)工作组(由疾病控制中心协调)和美国病理学家学会已经提出了用于保证质量ngs数据的标准和解释。例如,nex-stoct推荐了一系列关于ngs的后分析qc指标,其包括覆盖的深度和均匀性、转换/颠换比、碱基识别质量评分、定位质量及其他(gargis等人,2012)。迄今为止,已经公开许多方法用于变异识别。这些方法通常分为两类:仅肿瘤和匹配的肿瘤-正常。因为匹配的肿瘤-正常算法能够区分作为种系事件的生物突变或“真正”突变和作为体细胞事件的真正突变,所以它们是有吸引力的。然而,在临床实践中匹配样品测序更昂贵,往往不能获得。因此,拥有可以无需相应正常样品而进行、并仍实现高灵敏度和特异性的方法变得至关重要。一些团体已经建议使用来自同一组织、跨越多个种群成员或遗传相关对象的多个基因组序列的多个样品的同时评价来评价一个或更多个假设正确的概率(美国公开2012/0208706、2014/0057793和2014/0058681)。其他人已经建议使用为基因序列读取而计算的读取属性来评估读取结果(reads)(ep2602734a1)。还建议了通过样品dna的选择性验证区域验证ngs输出(ep2602734a1)。几个团体最近已经描述了专门开发用于dna样品中低水平的体细胞突变的方法(hadd等人,2013年,forshew等人,2012年,yost等人,2012),包括适应样品dna“噪音”的方法,例如在转换突变中的噪音升高(hadd等人,2013年)。然而,仍有改进测序算法和ngs变异识别算法的需求。技术实现要素:实施方案包括克服上述限制等的装置、系统、计算机可读介质、试剂盒以及方法。本公开重点在于将样品的可行模板计数并入后测序分析中样品的来减少样品输入需求,同时保持高灵敏度和阳性预测值(ppv)。其他改进包括靶向dna或rna基因座和使操作者在很短时间能够从提取的核酸进行到测序,包括质量控制步骤。此外,预测序质量控制与后测序分析的整合利用难以或不可能仅从测序数据推断的样品特异性细节、例如核酸的完整性或核酸输入到文库制备中的扩增拷贝数丰富了序列分析。本文公开的一些实施方案涉及一种方法,其包括定量包含核酸的样品中可行的模板计数;富集核酸的目标区域以创建用于测序的文库;从文库中生成序列数据,其中所述数据包括多个序列读取结果;基于一组序列读取结果将并入了样品的可行模板计数的基于计算机的变异识别模型用于识别目标区域序列的分析序列数据。预期的是变异识别模型可以通过能够访问测序数据并且执行在变异识别模型中包含的指令的计算设备来实现。在一些实施方案中,所述变异识别模型被配置成识别相对于参照序列的样品核酸中的一种或更多种序列变异。通过变异识别模型识别的序列变异包括但不限于单核苷酸变异、插入、缺失、多核苷酸取代、结构变异、基因组拷贝数变化、基因组重排、剪接变异和/或rna变异。变异可以代表种系突变、体细胞突变或两者。在一些实施方案中,一种或更多种序列变异与疾病状态和/或疾病倾向相关。预期本文公开的方法可以用于多种疾病或病症的诊断和/或预后或用于确定个体发展疾病或病症的倾向或可能性。疾病或病症可以包括具有遗传组分的那些疾病或病症和/或个体核酸序列信息在疾病或病症的诊断、预后或开方治疗中会有用的那些疾病或病症。还可以预期本文公开的方法可以用于预测个体的药物基因组学应答,例如对药物的耐药性、敏感性和/或毒性。在一些实施方案中,变异识别模型被配置为识别定量的目标特异性拷贝数变异。预期在本文公开的一些实施方案中,变异识别模型测序和/或变异识别的核酸可以来源于各种生物来源和/或合成来源。在一些实施方案中,核酸包括来自生物样品的dna、rna和/或总核酸。在一些实施方案中,核酸包括基因组dna。核酸可以从其而来的来源的非限制性实例包括:福尔马林固定石蜡包埋的组织、通过细针抽吸收集的组织、冷冻组织、血清、血浆、全血、循环肿瘤细胞、通过激光捕获显微切割收集的组织、芯针活组织检查、脑脊液、唾液、口腔拭子、粪便样品和尿。在一些实施方案中,样品中的核酸是异质的。这种异质核酸可以包括相对大量序列与样品中的其它分子相同、但在一些位置变化的核酸分子。包含异质核酸的组合物和样品可以例如由基因组dna样品中基因的不同等位基因在样品中的存在产生;由来源于不同源的样品中的核酸产生,例如当一些核酸来源于已经出现体细胞突变的细胞,而一些来源于没有出现相同体细胞突变的细胞;或者,在来自于存在于样品中的不同剪接变异的mrna的情况下。在一些实施方案中,样品中的核酸来自癌细胞和非癌细胞的混合物。在一些实施方案中,包含用于生成测序文库的核酸的样品具有低于约10000、9000、8000、7000、6000、5000、4000、3000、2000、1000、500、400、300、200、100或50的可行模板计数。在某些方面,可行的模板计数为10、20、30、40、50、100至150、200、300、400、500、1000、2000或更多,包括其间的所有值和范围。在一些实施方案中,定量可行模板计数包括进行定量的pcr分析。本文公开的一些实施方案涉及在样品中富集核酸的某些目标区域以产生测序文库。文库是包含进入测序反应的输入的核酸分子的集合。文库分子可以例如作为用于涉及文库分子的至少一部分复制的测序反应的模板。文库可以被设计为富集例如基因组的某些目标区域。即,相比非目标区域,文库可以具有目标区域的更多拷贝。在一些实施方案中,文库可以包括基本上仅目标区域,大部分非目标核酸已经通过纯化工艺除去。在一些实施方案中,富集核酸的目标区域来创建文库包括使用能够退火在目标区域延伸的一个或更多个dna引物对进行pcr反应。在一些实施方案中,pcr反应是多重反应。在一些实施方案中,富集核酸的目标区域包括进行捕获杂交过程。在本文公开的一些实施方案中,由文库生成序列数据包括平行地获取多个序列读取结果。这可以通过许多下一代测序平台来实现。在一些实施方案中,序列数据包括用于文库的每个部分的多个序列读取结果。在一些实施方案中,方法进一步包括将序列数据校正为参考序列。本文公开的一些实施例涉及基于一组序列读取结果将并入了样品可行模板计数的变异识别模型用于识别目标区域的序列。可以以多种不同的方式将可行模板计数并入变异识别模型,这将提高模型的准确性和实用性。在一些实施方案中,变异识别模型被配置为基于可行模板计数的值来调整序列假设为真实的概率。在一些实施方案中,变异识别模型被配置为如果变异模板计数低于阈值,则降低序列假设为真实的概率。在一些实施方案中,变异识别模型被配置为如果变异模板计数高于阈值,则升高序列假设为真实的概率。在一些实施方案中,变异识别模型被配置为基于可行模板计数的值来调整分配给模型特征的权重。在一些实施方案中,变异识别模型被配置为比较序列数据和参考序列。参考序列可以包括提供相对于可以识别变异的基线的历史或其他测序信息。在一些实施方案中,变异识别模型被配置为根据可行模板计数调整观察非参考碱基的先验概率。在一些实施方案中,变异识别模型被配置为并入可的行模板计数作为模型的特征。即,可行模板计数本身可以是变异识别模型的特征。在一些实施方案中,变异识别模型被配置为使用不同组的模型特征来识别样品中的序列变异,如果可行模板计数位于预定义区间内。在一些实施方案中,变异识别模型被配置为使用替代的分类器来识别核酸中的序列变异,如果可行模板计数位于预定义的区间内,例如,可行模板计数为10、20、30、40、50、100至150、200、300、400、500、1000、2000或更多,包括其间的所有值和范围。因此,可行模板计数本身不仅可以是变异识别模型的特征,其也可以影响模型的其他特征和模型考虑其他特征的方式。本文描述的实施方案利用本发明人的发现,将可行模板计数并入变异识别模型中使得模型比不这样做更精确和有用。在一些实施方案中,在本文所述的方法中使用的变异识别模型相对于不并入可行模板计数的相同变异识别模型,具有增加的阳性预测值(“ppv”)、减少的假阳性发生率和/或减少的假阴性发生率。在一些实施方案中,对于可行模板计数低于200、100、75、50或25和/或高于5、10、25、50、75或100、包括其间所有值和范围的样品,变异识别模型的ppv比不并入可行模板计数的相同变异识别模型至少高大约5%、10%、15%、20%、25%、30%、35%、40%、45%或50%。在一些实施方案中,变异识别模型对可行模板计数低于100的样品的灵敏度是不并入拷贝数的相同变异识别模型的90%或更高。在一些实施方案中,对于可行模板计数低于100、200、300、400或500的样品,或可行模板计数为10、20、30、40、50或60至100、200、400或500的样品,变异识别模型具有高于75%的ppv。在一些实施方案中,对于可行模板计数小于100、150或200的样品,或可行模板计数为10、20、30、40或50至100、150、200的样品,变异识别模型假阳性风险降低。在一些实施方案中,相对于不并入可行模板计数的相同变异识别模型,对于可行模板计数高于约1000、2000、3000、4000或5000的样品,或可行模板计数为1000、2000、3000、4000或5000至6000、7000、8000、9000或10000的样品,变异识别模型敏感性增加,并且对于那些样品ppv没有大量减少。在一些实施方案中,在本文公开的方法中使用的含核酸的样品包括来源于人对象的dna。如果核酸是在人对象体内产生的,则核酸是“来源于人对象的”。在一些实施方案中,上述方法还包括基于序列数据分析确定人对象是否具有疾病或疾病倾向。在一些实施方案中,疾病是癌症。在某些方面,方法通过使用本文中所述的变异识别方法评价来自对象的核酸样品中的变异,用于识别具有特定疾病或病症的对象,可能以积极或消极方式应答特定的疗法或治疗的对象。在一些实施方案中,该方法进一步包括基于序列数据的分析选择疾病治疗。在一些实施方案中,疾病治疗是施用抗癌疗法。抗癌疗法可以包括例如施用药物、化学疗法、放疗和/或手术。在一些实施方案中,方法还包括基于序列数据的分析选择不施用疾病治疗。在一些实施方案中,方法还包括基于序列数据的分析确定疾病治疗是否会对人对象显示需要治疗或禁用。还公开了改善被配置为通过分析序列数据进行序列识别的计算机执行的变异识别模型的方法,所述方法包括将输入样品的可行模板计数并入序列数据的模型分析中来改进模型。在一些实施方案中,可行模板计数值基于定量pcr分析。在一些实施方案中,定量pcr分析测量了通过模型分析的序列数据来源的文库中与pcr扩增子有相似大小的dna片段的扩增。在一些实施方案中,将可行模板计数并入到测序数据的模型分析包括基于可行模板计数的值配置模型以调整序列假设为真实的概率。在一些实施方案中,如果变异模板计数低于阈值,例如,100、50、40、30、20或10,将可行模板计数并入到测序数据的模型分析中包括配置模型以降低序列假设为真实的概率。在一些实施方案中,如果变异模板计数高于阈值(例如,50、100或200),将可行模板计数并入到测序数据的模型分析中包括配置模型以升高序列假设为真实的概率。在一些实施方案中,将可行模板计数并入到测序数据的模型分析中包括基于可行模板计数的值配置模型以调整分配给模型特征的权重。在一些实施方案中,将可行模板计数并入到测序数据的模型分析中包括根据可行模板计数配置模型以调整观察非参考碱基的先验概率。在一些实施方案中,将可行模板计数并入到测序数据的模型分析中包括配置模型以并入可行模板计数作为模型特征。在一些实施方案中,如果可行模板计数位于预定义的区间内,将可行模板计数并入到测序数据的模型分析中包括配置模型以使用不同组的模型特征来识别样品中的序列变异。在一些实施方案中,如果可行模板计数位于预定义的区间内,将可行模板计数并入到测序数据的模型分析中包括配置模型以使用替代的分类器来识别序列变异。在一些实施方案中,改进的变异识别模型相对于改进前的变异识别模型具有增加的ppv、减少的假阳性发生率和/或减少的假阴性发生率。在一些实施方案中,对于拷贝数低于100、75、50或25;或5、10、15、或20至25、50、75、100的输入dna,改进的变异识别模型的ppv比改进前的变异识别模型高至少约5%、10%、15%、20%、25%、30%、35%、40%、45%、或50%。在一些实施方案中,对于可行模板计数小于100的输入样品,改进的变异识别模型的灵敏度是改进前的变异识别模型的灵敏度的90%或更高。在一些实施方案中,对于可行模板计数低于100、200、300、400、或500;或可行模板计数为5、15、25、50或75至100、200、300、400、或500的输入等分试样,改进的变异识别模型具有高于75%的ppv。在一些实施方案中,对于可行模板计数小于100、150或200的输入等分试样,改进的变异识别模型相对于改进前的模型假阳性风险降低。在一些实施方案中,方法还包括使用一组已知变异和来源于具有变化的可行模板计数值的输入样品的测序数据来训练模型,输入样品包括具有少于约100个功能性dna拷贝的样品和具有大于约500个功能性dna拷贝的样品。还公开了一种非暂时性机器可读存储介质,其包括当由计算设备执行时引起计算设备进行至少以下步骤的指令:访问与核酸分子文库相关的序列数据,其中所述文库是由核酸输入样品生成的;和分析序列数据以通过考虑与输入样品相关的可行模板计数识别序列变异。访问序列数据可以包括例如获得序列数据和/或接收序列数据。在一些实施方案中,文库包含通过pcr和/或捕获杂交由核酸输入样品富集的核酸分子。在一些实施方案中,经富集的核酸分子与疾病状态、疾病倾向和/或对药物治疗的药物基因组应答相关。在一些实施方案中,可行模板计数已经通过定量pcr分析计算。在一些实施方案中,核酸输入样品来源于选自以下中的一种或更多种的生物样品:福尔马林固定石蜡包埋组织、通过细针抽吸收集的组织、冷冻组织、血清、血浆、全血、循环肿瘤细胞、通过激光捕获显微切割收集的组织、芯针活组织检查、脑脊液、唾液、口腔拭子、粪便样品和尿。在一些实施方案中,输入核酸包括来自生物样品的dna、rna和/或总核酸。在一些实施方案中,输入核酸包括基因组dna。在一些实施方案中,考虑与输入样品相关的可行模板计数包括基于可行模板计数值调整序列假设为真实的概率。在一些实施方案中,如果变异模板计数低于阈值,考虑与输入样品相关的可行模板计数包括降低序列假设为真实的概率。在一些实施方案中,考虑如果变异模板计数高于阈值,与输入样品相关的可行模板计数包括升高序列假设为真实的概率。在某些方面,阈值可以是预先确定的数或经计算的数。在一些实施方案中,基于可行模板计数的值,考虑与输入样品相关的可行模板计数包括调整分配给变异识别模型的特征的权重。在一些实施方案中,考虑与输入样品相关的可行模板计数包括根据可行模板计数调整观察非参考碱基的先验概率。在一些实施方案中,考虑与输入样品相关的可行模板计数包括并入可行模板计数作为模型的特征。在一些实施方案中,如果可行模板计数位于预定义的区间内,考虑与输入样品相关的可行模板计数包括使用不同组的模型特征来识别样品中的序列变异。在一些实施方案中,如果可行模板计数位于预定义的区间内,考虑与输入样品相关的可行模板计数包括使用替代的分类器以识别序列变异。还公开了一种用于确定核酸序列的试剂盒,其包括:(a)定量pcr试剂组,其能够用于确定样品中核酸的可行模板计数;(b)多重pcr试剂组,其能够用于扩增样品中的多个目标区域,并产生用于测序的核酸分子文库;(c)标记pcr试剂组,其能够用于将序列附加到文库中的核酸分子上;(d),能够用于纯化和/或归一化文库中的核酸分子用于在测序前进一步扩增的试剂组;(e)非暂时性机器可读的存储介质,其包括当通过计算设备执行时引起计算设备通过进行至少以下步骤识别序列变异的指令:(i)访问或接收与核酸分子文库相关的序列数据;和(ii)分析序列数据以通过考虑与样品相关的可行模板计数来识别序列变异。在一些实施方案中,定量pcr试剂组包括能够用于使缓冲剂适合于定量pcr的母料混合物。在一些实施方案中,定量pcr试剂组包括用于扩增样品中核酸的区域或片段的引物。在一些实施例中,多重pcr试剂组包括配置为扩增至少5、10、15、20、25、30、35、40、45或50个与疾病状态或疾病倾向相关的基因组区域的引物。在一些实施方案中,基因组区域覆盖至少50、100、200、300、400、500、600、700或800个与疾病状态或疾病倾向相关的基因座。在一些实施方案中,疾病是癌症。在一些实施方案中,基于可行模板计数值,考虑与样品相关的可行模板计数包括调节序列假设为真实的概率。在一些实施方案中,如果变异模板计数低于阈值,考虑与样品相关的可行模板计数包括降低序列假设为真实的概率。在一些实施方案中,如果变异模板计数高于阈值,考虑与样品相关的可行模板计数包括升高序列假设为真实的概率。在一些实施方案中,基于可行模板计数值,考虑与样品相关的可行模板计数包括调整分配给变异识别模型的特征的权重。在一些实施方案中,考虑与样品相关的可行模板计数包括根据可行模板计数调整观察非参考碱基的先验概率。在一些实施方案中,考虑与输入样品相关的可行模板计数包括并入可行模板计数作为模型的特征。在一些实施方案中,如果可行模板计数位于预定义的区间内,考虑与样品相关的可行模板计数包括使用不同组的模型特征来识别样品中的序列变异。在一些实施方案中,如果可行模板计数位于预定义的区间内,与样品相关的可行模板计数包括使用替代的分类器以识别序列变异。还公开了一种在基因组dna样品中识别变异的方法,其包括:(a)进行定量pcr分析以确定包含核酸的样品中可行模板浓度;(b)使用可行模板浓度以计算样品的等分试样中的可行模板计数;(c)使用等分试样作为模板进行pcr反应以产生富集感兴趣的核酸片段的文库;(d)由文库产生序列数据;和(e)使用并入可行模板计数的基于计算机的变异识别模型分析序列数据以识别基因组dna中的序列变异,其中并入可行模板计数包括配置模型以进行以下的一个或更多个步骤:基于可行模板计数值调整序列假设为真实的概率;如果变异模板计数低于阈值,降低序列假设为真实的概率;如果变异模板计数高于阈值,升高序列假设为真实的概率;基于可行模板计数值调整分配给模型特征的权重;根据可行模板计数调整观察非参考碱基的先验概率;并入可行模板计数作为模型的特征;如果可行模板计数位于预定义的区间内,识别样品中的序列变异;和/或如果可行模板计数位于预定义的区间内,使用替代的分类器以识别核酸中的序列变异。还公开了一种提高核酸样品的变异识别质量的方法,其包括:(i)确定待测序样品中功能性拷贝的量,(ii)基于样品中功能性拷贝的量,确定待用于测序中的样品量。在一些实施方案中,功能性拷贝为rna功能性拷贝。在一些实施方案中,待用于测序中的样品的确定量包括至少100、200、300、或400个功能性拷贝。在一些实施方案中,产生序列数据可以包括平行地获得多个序列读取结果。这可以通过例如以下方式实现,使用下一代测序(ngs)平台,其包括但不限于来自illumina、pgm的miseq、hiseq或nextseq仪器,或来自thermofisher的proton仪器,由roche/pacificbiosciences、completegenomics、oxfordnanopore、biorad/gnubio、genia、stratos、noblegen、lasergen和nabsys提供的其他平台。在一些实施方案中,样品包含rna,方法包括识别样品中rna中的变异。这样的实施方案可以包括在定量pcr步骤前的反转录步骤,进行pcr以创建文库的步骤,或两者。在本文描述的一些实施方案中,变异识别模型被配置为基于可行模板计数调整变异假设的概率。可行模板计数可以用作评价变异假设的模型特征。此外或任选地,可行模板计数可用于调整在评价变异假设中使用的另一个模型特征的权重或评分。实施方案还包括但不限于方法、试剂盒、装置、系统和计算机可读介质,其用于提高识别来自患者的遗传变异的分析的准确性和/或灵敏度、基于识别一种或更多种遗传变异诊断患者患有疾病或病症、基于测序多个标记物诊断患者、在具有低丰度的高质量遗传材料的样品中的识别遗传变异、减少遗传变异的假阳性判定、减少遗传变异的假阴性判定、使用改善变异识别的算法用于以较高的精确度确定一个或更多个序列是否变异、使用变异识别模型以改善诊断、或确定生物样品中潜在变异的序列。在各种实施方案中,基因测序仪用于识别遗传变异,使用改善输出的经训练的算法以考虑在被测序样品中是否有足够数量的良好核酸模板可用,来评价测序输出。在某些实施方案中,系统包括计算机硬件以运行提高变异识别的算法。任何这些实施方案可以与本公开所描述的步骤和/或组件一起使用。在某些实施方案中,有基于确定患者在从患者获得的核酸样品中是否具有遗传变异来诊断患者的方法,其包括:分析核酸样品的至少一部分以确定在涉及经扩增的核酸分子的测序反应中可使用的核酸模板数;扩增样品中的核酸分子;在包括与疾病或病症相关的潜在变异的一个或更多个区域测序经扩增的核酸分子;和使用算法以评价来自序列经扩增的核酸分子的数据。如果患者被识别具有表明特定治疗方案的一种或更多种基因序列,在某些实施方案中,治疗患者与一种或更多种基因序列相关的疾病或病症。预期本说明书中所讨论的任何实施方案可以对于本发明的任何方法、系统、试剂盒、计算机可读介质或装置来实施,反之亦然。此外,本发明的装置可以用于实现本发明的方法。术语“大约”或“约”定义为如本领域普通技术人员所理解的接近于,在一个非限制性实施方案中该术语定义为在10%以内、优选在5%以内、更优选在1%以内、最优选在0.5%以内。术语“基本上”及其变体定义为如本领域普通技术人员所理解的大部分但不必全部地为指定的事物,在一个非限定性实施方案中,基本上涉及的范围为在10%以内、在5%以内、在1%以内或在0.5%以内。术语“抑制”或“减少”或这些术语的任何变体包括实现所期望结果的任何可测量的减少或完全的抑制或减少。术语“促进”或“增加”或这些术语的任何变体包括实现所期望结果的任何可测量的核酸、蛋白质或分子的增加或产生。如本说明书和/或权利要求中所使用的术语,术语“可行的的”表示足以实现期望的、预期的或想要的结果。当在权利要求和/或说明书中与术语“包含”一起使用时,要素前面不使用数量词可以表示“一个”,但是其也符合“一个或更多个”、“至少一个”和“一个或多于一个”的意思。如本说明书和权利要求所使用的,单词“包含”、“具有”、“包括”或“含有”是包括性的或开放式的,并且不排除另外的、未列举的元素或方法步骤。使用的装置和方法可以“包含”贯穿本说明书所公开的任何成分或步骤、“基本由其组成”或“由其组成”。“变异”是某物与相同事物的其他形式或与标准在一些方面不同的形式或版本。当用于指核酸序列时,“变异”是与相同核酸的其他形式或标准核酸在一些方面不同的核酸。非限制性实例是单核苷酸多态性(snp);单核苷酸变异(snv);复杂的碱基变化,如多核苷酸取代;结构变异、基因组拷贝数变化和重排、定量拷贝数估计和/或其组合。与变异不同的标准或相同核酸的其它形式可以是但不限于生物核酸、非生物核酸、合成核酸、植物核酸、动物核酸、真菌核酸、原核生物核酸、人核酸、正常组织核酸、癌组织核酸、患病组织核酸、先前核酸、来自遗传相关生物体或家族成员的核酸、代表在种群中发现的通用或特定核酸的核酸、人工核酸、来自标准品的核酸、来自文库中另一个样品的核酸、来自相同样品的核酸和/或其组合。“变体识别模型”或“变体识别器”是一组指令,通过其计算机分析核酸测序数据以识别目标核酸分子中的序列和/或变异(即,以表明序列或表明在目标核酸分子特定位置的序列是否相对于参照序列不同或没有不同)。在一些实施方案中,变异识别模型(1)评估了样品中的核酸分子具有序列变异(即,偏离参考序列)的概率或可能性,和(2)提供信息和/或生成关于在样品中可能存在或不存在的一个或更多个变异,如果存在的话,这些变异在样品中的可能频率的报告。在一些实施方案中,变异识别模型表明序列或变异识别的误差的确定性或概率,其包括,在一些实施方案中表明在一个位置没有变异的误差的确定性或概率。如果第一分子为第二dna分子大小的约85至115%,则第一dna分子是与第二dna分子相似大小的。“可行模板”是一种核酸,其是pcr可扩增的,通过任意酶过程可扩增的,和/或通过任意蛋白质或蛋白质部分可操纵的,且其来自含有待通过一种或更多种化学或物理测试分析的核酸的样品。“可行模板浓度”是每体积单位的可行模板数。在一些实施方案中,其可使用定量pcr系统如qpcrdnaqc分析来确定。在一些实施方案中,其可以使用显示可行模板计数的任何其他方法确定,所述方法包括但不限于实时pcr、数字pcr或等温扩增方法。“可行模板计数”是包含样品核酸的等分试样中的可行模板的绝对数。可以计算等分试样的可行模板计数的一种方式是将样品的可行模板浓度乘以从样品取出的等分试样的体积。可行模板计数还可以通过显示包含核酸的组合物中可行模板的量的任意其他方式来计算。在一些实施方案中,在进行序列识别和/或识别序列变异中变异识别模型考虑可行模板计数。本发明的其它目的、特征和优点通过以下详细的描述会变得明显。然而,应理解详细的描述和实施例在表明本发明的具体实施方案时仅以举例说明给出。另外,期望通过该详细描述,本发明的精神和范围内的变化和修改对于本领域技术人员将会变得明显。附图说明以下附图形成本说明书的一部分,并被包含以进一步证实本发明的特定方面。通过参照一个或更多个这些附图结合本文所提供的具体实施方案的详细说明可以更好地理解本发明。图1-工作流程中显示了预期方法或试剂盒的一个实施方案的一般结构和元素。图2a和2b-(a)预期方法或试剂盒的实施方案的组件整合了具有样品定量的基于pcr的富集工作流程的元素与生物信息学。(b)pancancerdna面板。图3a和3b-(a)dnaqc方法学概述。(b)用于rna和dna靶的全部整合的工作流程的概述,包括qc试剂、ngs试剂、其他工作流程组件和可行变体识别器。在一个实施方案中,ngs系统是从qc到信息学改进的工作流程,其能够同时定量来自与低输入、低质量样品分离的总核酸(tna)的dna点突变、插入和缺失、结构变异、rna表达和基因融合。作为非限制性实例,可以使用从样品分离的总核酸定量功能性dna和rna的新型qpcr分析来进行靶向ngsqc。可以使用靶向ngs试剂进行基于pcr的目标富集,并在(illumina)上测序。可以使用ngs报告器分析文库序列,ngs报告器(reporter)是直接并入预分析qc信息以提高变异识别、融合检测和rna定量的准确性的生物信息学分析套件。图4-预期的方法或试剂盒的实施方案,其能够定量和富集来自从人组织或细胞系纯化的dna的几种基因的癌症相关变异。该试剂盒或方法支持使用测序仪器(本文展示的illuminamiseq仪器)的多重下一代测序分析。该试剂盒或方法包括用于确定qfi分析评分和抑制的组件,使用本地整合的生物信息管线和伴随数据的可视化工具分析序列文件例如fastq用于识别碱基取代突变和小插入/缺失的profile软件。图5-应用试剂盒以确定一组临床核酸分离物的qfi分析评分和抑制曲线。图6a和6b-(a)在方法和/或试剂盒实施方案中预期的pcr的2个步骤的实例:i)利用连接到每个引物的共同序列的基因特异性扩增;ii)第二pcr附加仪器-特异性连接头(adapters)和索引编码(indexcodes)被添加到pcr产物。将来自各个样品的产物混池(pooled),然后聚集到流动池(flowcell)上。在成像之后,使用索引编码将各个测序读取结果分配到它们各自的文库。(b)显示了双索引编码(dualindexcodes)的实例(具有ilmn接头、特定编码和cs1/cs2区域)。图7–母料混合物(mastermix)设置(setup):引物混合物(3545-1)-92个引物对,2×pcr母料混合物(3469-1)(与ngs核心试剂相同),固定体积为4μl的样品;标记pcr的“无母料混合物”设置-寡核苷酸作为预混合物,2×母料混合物(3469-1)和基因特异性产物的等分试样。图8a和8b-使用(a)操作者1、2和3(分别为3.9%、5.3%、6.5%)和(b)石蜡包埋样品的扩增子产量、总覆盖率和操作者之间的变化性突出了面板的性能。图9-当用缺少可行模板信息的变体识别器时,dnaqc利用有限的可行模板分子的细胞(cp#)显示了提高的假阳性突变识别。图10a和10b-有限的功能性拷贝极大增加了假阳性(右格)的风险,并限制了灵敏度(左格)。可行识别器在跨越功能性拷贝输入的整个范围内显示了一致的性能。与没有考虑输入拷贝数的识别器相比,asuragen变异识别器显示了在低功能性模板拷贝时假阳性识别的抑制,同时保持对已知阳性brafv600e(a)和krasg12v(b)的高灵敏度。这些样品未在训练模型中使用。图11-模型建立输入和策略的略图。图12-对推定的种系和推定的体细胞变异评估性能。显示的是每组中百分比变异的分布,其说明推定的种系变异遵循预期的生物模式分布,而推定的体细胞变异在整个范围内是难以分辨的,其对于低百分比变异(<25%)具有严重偏好。图13-各种当前阶段变异识别器(variantcaller)的等位基因频率的灵敏度,如http://genomemedicine.com/content/5/10/91/中所评估的。图14-可行的识别器(-enabledcaller)在1%至100%变异改善ppv,提供了相对于基线在相同范围内的等效或更好的灵敏度。图15-可行的识别器在输入的整个范围是灵敏的。可行识别器特别有利于低输入样品,其相对于低于100个拷贝的基线模型使ppv增加了50%。描述了推定的体细胞变异的性能。图16-推定的种系变异的性能表。基线模型和可行的模型在此数据集上产生等同的结果。图17-在超过600个ffpe样品的群体中,使用10ng输入,大于27%的样品会含有<100个dna的功能性拷贝。变异识别器相对于基线和其他现存的变异识别器极大地降低了该集合中的假阳性风险。图18-识别器显示极高的分析灵敏度,其正确地识别了少到1.7个突变体拷贝。图19-qc显示了用靶向erbb2基因的面板测序的不同质量的51个ffpe样品,可使用的测序读取结果(y轴)%与输入到测序反应中的功能性拷贝(x轴)之间的关系。图20-使用识别器下一代测序(ngscnv)和微滴式数字pcr(biorad,sep25)的拷贝数变化检测的比较。图21-样品内相对扩增效率的标准偏差。随着dna质量评分(qfi)减少,相对效率差异加剧,导致从预期基线的偏离增加。图22-相比基于qpcr的方法,通过基于ngs的方法估计任何大小范围的功能性dna百分比(brisco等人,2010)。图23-通过增加文库质量输入可以拯救较低质量的样品(通过rna功能性拷贝分析分级)。图24-rna功能性拷贝预测了两个独立靶向rna-seq面板的靶向测序数据质量:40个靶mrna表达面板(左)和50个靶基因融合面板(右)。用少于100个可行rna模板分子制备的文库显示了对预期目标减少的比对率(mappingrate)和对两个面板的引物二聚体形成的升高速率。图25-rna功能性拷贝与由ngs产生的目标读取结果相关。由100ng到0.01ng的完整tna输入滴定的三个tna显示了功能性rna模板拷贝和对靶比对率(targetmappingrate)的后测序比质量输入和在靶比对率上有更强相关性。具体实施方式如上所述,本发明的一个独特方面是在测序结果的后测序分析中并入样品的可行模板计数。这允许了减少的样品输入要求,同时保留了高灵敏度和阳性预测值(ppv)的益处,靶向dna基因座和rna基因座两者,使得操作者在包括质量控制步骤的短时间内测序提取的核酸。此外,预测序质量控制与后测序分析的整合利用难以或不可能仅从测序数据中推断的样品特异性细节、例如核酸的完整性或输入到文库制备中的可扩增拷贝数,丰富了序列分析。确定样品中核酸的功能性拷贝数或可行模板计数的百分比或量可以用于确定满足进行分子分析的最小核酸要求所需的样品量(sah等人,2013,wo公开2013/159145)。迄今为止,已经公开了用于确定核酸的可行模板计数的百分比或量或损伤频率的几种方法(sah等人,brisco等人,2010,brisco等人,2011,us公开2012/0322058,wo公开2003/159145)。例如,近期已经描述了pcr定量分析的结果,其被称为定量的功能性指数pcr或qfi-pcr,其通过测量能够进行pcr扩增的dna模板的数和百分比可以用于计算用于分子分析如靶向pcr富集的样品输入的最小量(sah等人,2013)。使用实验室研发和可商购获得的富集程序和随后的ngs,这种洞察可以减少变异识别中假阳性和假阴性的风险。因此,基于qfi-pcr的预分析步骤的整合提供了极为改善的方法以确保ngs数据解释的准确性,其不仅用于在ngs之前评估ffpedna,还用于依赖pcr扩增的其他分析。因此,考虑dna质量进行解释,dna不良样品的严格和定量表征对于确保由功能性dna模板的足够拷贝产生的结果是必不可少的,并且其可以支持可靠的突变识别。基于由dna模板的不充分扩增造成的测序结果的误导性诊断后果是严重的,并且可能导致通过未识别可行性突变或基于假阳性结果开出错误治疗的不恰当的患者治疗。这种错误也可能破坏与癌症药物研发相关的回顾性生物标志物关联研究。然而,即使使用前述的qfi-pcr以确定基于pcr的分子分析中所需的样品dna的合适量,也没有解决低质量样品的ngs序列识别中的所有挑战。以下小节会更详细地描述本发明的非限制性方面。a.核酸样品预期本文描述的实施方案可以包括任何类型的核酸,其包括但不限于dna、rna、单链核酸、双链核酸、异质核酸、均质核酸、来自正常细胞的核酸、来自癌细胞的核酸、来自正常细胞和癌细胞混合物的核酸、和/或其组合。核酸源的非限制性实例包括生物源、非生物源、合成源、临床或非临床源、血浆/血清、新鲜组织、冷冻组织、循环肿瘤细胞、激光捕获显微解剖(lcm)组织活检、芯针活组织检查、细针抽吸(fna)组织、全血、脑脊液(csf)、唾液、口腔拭子、粪便样品、尿、肿瘤、福尔马林固定石蜡包埋组织(ffpe)、和/或其组合。在一些方面,核酸样品可以包含在含有核酸的样品的等分试样或提取物中。b.可行模板计数的确定预期实施方案可以包括用于确定可行模板计数的所有类型的方法和装置。用于确定可行模板计数的实施方案的非限制性实例包括qfi-pcr、定量pcr、实时pcr、数字pcr、显示可扩增拷贝数的其他基于pcr的方法、非pcr方法,其包括但不限于等温扩增、滚环扩增或相似方法和/或其组合。另外的非限制性实例包括美国公开2014/0051595,sah等人,2013,brisco等人,2010,brisco等人,2011,美国公开2012/0322058和wo公开2013/159145中描述的方法和装置。c.测序文库的创建预期本发明的方法和装置可以包括用于创建测序文库的所有类型的方法和装置。非限制性实例包括通过基于pcr的方法、基于多重pcr的方法、基于捕获杂交的方法和/或其组合的任何方式富集目标区域。还预期文库可以含有:一个或更多个感兴趣的次基因组区域、一个或更多个感兴趣的扩增区域;和/或与任意疾病、病症、状态、药物基因组学应答(例如耐药性、敏感性和/或毒性)、倾向性相关的一个或更多个感兴趣的区域和/或其组合。d.测序数据的生成预期本发明的方法和装置可以包括用于生成测序数据的所有类型的方法和装置。非限制性实例包括基于pcr和非基于pcr的方法,miseq仪器、hiseq仪器、nextseq仪器、pgm仪器、proton仪器、roche/pacbio平台、oxfordnanopore平台、completegenomics平台、genia平台、stratos平台、biorad/gnubio平台、nabsys平台等。还预期测序数据可以包括对于文库的每个部分的一个或更多个序列读取结果和/或对于文库的一个或更多个部分无读取结果。还预期测序平台、仪器或机器可以被配置为串联或平行地测序单一或多个文库片段。e.变异识别模型变异识别模型可以被配置为具有用于确定测序数据是否表明样品中变异的可能存在的各种指令。作为实例,测序读取结果与参考序列比对可以表明单核苷酸变异(snv)存在于输入dna中的给定位置。这导致snv存在于该位置的“变异假设”。为了评估输入dna实际上在该位置具有snv的可能性(即变异假设是真实的),变异识别模型可以被配置为考虑序列数据的各个方面作为模型特征、协变量和/或进行评估的分类器。一个这样的标准可以是同样表明该相同snv的测序读取结果的比例。模型可以指令计算机如果比例低,则样品中实际存在snv的概率应该降低。作为另一个实例,模型可以被配置为考虑来自互补链的测序读取结果是否显示相同的snv,并相应地调整snv存在于输入dna中的概率。变异识别模型可以包括用于评估变异概率的任何数量的模型特征、协变量和/或分类器。可能的变异的最终列表及其频率是将所有模型指令应用到来源于原始测序数据的所有变异假设的结果。预期本发明的方法和装置可以包括所有类型的变异识别模型中的一种或更多种。模型的非限制性实例可以包括线性模型、线性判别分析(lda)、对角线线性判别分析(dlda)、随机森林、支持向量机(svm)、逻辑回归、泊松回归、贝叶斯网络和其他图形模型、决策树、提升树、k均值聚类和神经网络、隐马尔可夫模型(hmm)和/或其组合。具体的,变异识别模型的非限制性实例包括:surascore-一种基于泊松的模型,其通过泊松测试计算根据质量评分给出的变异概率,用于质量评分>q15的碱基。在该方案中由低质量测序产生的假变异权重下降,并且其可能被归类为阴性,而来自高质量测序数据的变异可以以高灵敏度和良好的特异性被识别。该模型适用于低频率突变体的高灵敏度检测。surascorebb-一种基于β二项式的基因分型模型。该模型适用于种系snp的准确和敏感检测,并且使用来源于历史测序数据的先验概率分布信息。预期变异识别模型可以以任何方式将可行模板计数并入。将可行模板计数并入变异识别模型的方法的非限制性实例可以包括以下方法:模型降低、升高,其基于可行模板计数包括、不包括或修改样品中存在的一个或更多个变异的概率;模型降低、升高,其包括、不包括或修改一个或更多个模型特征、协变量和/或分类器的权重或用途;和/或模型降低、升高,其包括、不包括或修改在识别序列中使用的一个或更多个序列读取结果。在变异识别模型中并入可行模板计数的进一步具体的非限制性方法可以包括以下方法:(1)直接包括可行模板计数的数目和/或“qfi”(dna质量评分),其可以包括但不限于:(a)功能性拷贝样品-通过可行模板计数分析直接报告的功能性拷贝数;(b)功能性拷贝面板(panel)-使用预测来自qfi、面板的中值扩增子大小和功能性拷贝样品的信息的模型调整用于测序面板(sequencingpanel)的中值扩增子大小的样品的可行模板计数的数目;(c)功能性拷贝扩增子-基于覆盖位置的扩增子长度在每个位置碱基上调整样品的功能性拷贝数,其可以利用基于qfi和功能性拷贝样品预测功能性拷贝的模型。(2)以拷贝依赖的方式修改其他评分标准。这种类型的特征可以是但不限于基于评分指标在序列读取结果之间假定统计学独立性的知识,但是当不充足的材料被投入初始反应中用于生成文库时,这种假设失效。在这种情况下,读取结果之间存在高度相互依赖性。这些特征一般计算如下:拷贝调整评分=评分/最大((覆盖/功能性拷贝样品),1);其中功能性拷贝样品可以用功能性拷贝面板和功能性拷贝扩增子替代,以分别产生为面板中的扩增子大小或单个扩增子大小调整的指标。预期变体识别模型可以使用一个或更多个可行模板计数阈值或可行模板计数范围阈值。可行模板计数阈值的非限制性实例包括总核酸含量或可行模板计数的拷贝或数目的百分比,例如:总核酸的0.0001%、0.0002%、0.0003%、0.0004%、0.0005%、0.0006%、0.0007%、0.0008%、0.0009%、0.0010%、0.0011%、0.0012%、0.0013%、0.0014%、0.0015%、0.0016%、0.0017%、0.0018%、0.0019%、0.0020%、0.0021%、0.0022%、0.0023%、0.0024%、0.0025%、0.0026%、0.0027%、0.0028%、0.0029%、0.0030%、0.0031%、0.0032%、0.0033%、0.0034%、0.0035%、0.0036%、0.0037%、0.0038%、0.0039%、0.0040%、0.0041%、0.0042%、0.0043%、0.0044%、0.0045%、0.0046%、0.0047%、0.0048%、0.0049%、0.0050%、0.0051%、0.0052%、0.0053%、0.0054%、0.0055%、0.0056%、0.0057%、0.0058%、0.0059%、0.0060%、0.0061%、0.0062%、0.0063%、0.0064%、0.0065%、0.0066%、0.0067%、0.0068%、0.0069%、0.0070%、0.0071%、0.0072%、0.0073%、0.0074%、0.0075%、0.0076%、0.0077%、0.0078%、0.0079%、0.0080%、0.0081%、0.0082%、0.0083%、0.0084%、0.0085%、0.0086%、0.0087%、0.0088%、0.0089%、0.0090%、0.0091%、0.0092%、0.0093%、0.0094%、0.0095%、0.0096%、0.0097%、0.0098%、0.0099%、0.0100%、0.0200%、0.0250%、0.0275%、0.0300%、0.0325%、0.0350%、0.0375%、0.0400%、0.0425%、0.0450%、0.0475%、0.0500%、0.0525%、0.0550%、0.0575%、0.0600%、0.0625%、0.0650%、0.0675%、0.0700%、0.0725%、0.0750%、0.0775%、0.0800%、0.0825%、0.0850%、0.0875%、0.0900%、0.0925%、0.0950%、0.0975%、0.1000%、0.1250%、0.1500%、0.1750%、0.2000%、0.2250%、0.2500%、0.2750%、0.3000%、0.3250%、0.3500%、0.3750%、0.4000%、0.4250%、0.4500%、0.4750%、0.5000%、0.5250%、0.5500%、0.5750%、0.6000%、0.6250%、0.6500%、0.6750%、0.7000%、0.7250%、0.7500%、0.7750%、0.8000%、0.8250%、0.8500%、0.8750%、0.9000%、0.9250%、0.9500%、0.9750%、1.0%、1.1%、1.2%、1.3%、1.4%、1.5%、1.6%、1.7%、1.8%、1.9%、2.0%、2.1%、2.2%、2.3%、2.4%、2.5%、2.6%、2.7%、2.8%、2.9%、3.0%、3.1%、3.2%、3.3%、3.4%、3.5%、3.6%、3.7%、3.8%、3.9%、4.0%、4.1%、4.2%、4.3%、4.4%、4.5%、4.6%、4.7%、4.8%、4.9%、5.0%、5.1%、5.2%、5.3%、5.4%、5.5%、5.6%、5.7%、5.8%、5.9%、6.0%、6.1%、6.2%、6.3%、6.4%、6.5%、6.6%、6.7%、6.8%、6.9%、7.0%、7.1%、7.2%、7.3%、7.4%、7.5%、7.6%、7.7%、7.8%、7.9%、8.0%、8.1%、8.2%、8.3%、8.4%、8.5%、8.6%、8.7%、8.8%、8.9%、9.0%、9.1%、9.2%、9.3%、9.4%、9.5%、9.6%、9.7%、9.8%、9.9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%、23%、24%、25%、26%、27%、28%、29%、30%、35%、40%、45%、50%、60%、65%、70%、75%、80%、85%、90%、95%、99%等,或其可得出的任何百分比或范围;或0、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000、2000、3000、4000、5000、6000、7000、8000、9000、10000、20000、30000、40000、50000、60000、70000、80000、90000、100000、200000、300000、400000、500000、600000、700000、800000、900000、1000000、2000000、3000000、4000000、5000000、10000000等,可行模板计数或其中可得出的任何数目或范围和/或其组合。还预期可以训练变异识别模型。变异识别模型可以在来源于任何输入核酸的任何数据组上训练。预期来源于输入核酸的变异或序列数据可以具有或可以不具有:拷贝数的统一、变化或组合;可行模板计数的统一、变化或组合;和/或变异识别模型考虑的任何其他因素的统一、变化或组合。预期变异识别模型的全部或部分可以在一个或更多个计算机可读存储介质上存储或可以不在一个或更多个计算机可读存储介质上存储。还预期一个或更多个计算机可读存储介质可以通过本地处理器、远程处理器、通过因特网接口和/或其任意组合执行或不执行。f.模型特征、协变量和分类器预期本发明的方法和装置可以包括所有类型的模型特征、协变量和/或分类器。模型特征和协变量的非限制性实例可以包括以下中的一种或更多种:评分指标、百分比变异、质量评分、覆盖深度、来源于历史数据的β基因型分型、功能性拷贝输入、可行模板计数、在感兴趣的碱基上游或下游定义的窗口中鸟嘌呤(g)和/或胞嘧啶(c)的百分比、在感兴趣碱基上游或下游定义的窗口中观察到的最长均聚物、观察突变体和接近读取结果末端之间关联有多强的测量、读取结果内碱基所在的位置和在碱基处观察到突变的可能性之间关联有多强的测量、功能性拷贝或所使用的可行模板分析的形式、功能性拷贝或使用的可行模板分析(tna或dna)中的输入类型、跨越所有假设的百分比变异的第95百分位、相对于中值样品覆盖讨论的碱基的覆盖、测序讨论的碱基次数、从考虑的位置的3'方向上移除一个碱基对的碱基识别、在考虑的位置的3'方向上10个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的3'方向上10个碱基的最长均聚物延伸、从考虑的位置的3'方向上15个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的3'方向上15个碱基的最长均聚物延伸、从考虑的位置的3'方向上两个碱基对的碱基识别、从考虑的位置的3'方向上20个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的3'方向上20个碱基的最长均聚物延伸、从考虑的位置的3'方向上三个碱基对的碱基识别、从考虑的位置的3'方向上5个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的3'方向上5个碱基的最长均聚物延伸、从读取结果边缘的三个位置内出现的变异数、从读取结果边缘的三个位置内出现的碱基总数、百分比变异的假设特定的第95百分位、假设(a>c、g>t等)、变异的全球人口次要等位基因频率、在位置的中值qscore、在该位置的qscore的三平均值(qscore的第25百分位、第50百分位和第75百分位的平均值)、覆盖位置的配对总数、从考虑的位置的5'方向上一个碱基对的碱基识别、从考虑的位置的5'方向上10个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的5'方向上10个碱基的最长均聚物延伸、从考虑的位置的5'方向上15个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的5'方向上15个碱基的最长均聚物延伸、从考虑的位置的5'方向上两个碱基对的碱基识别、从考虑的位置的5'方向上20个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的5'方向上20个碱基的最长均聚物延伸、从考虑的位置的5'方向上三个碱基对的碱基识别、从考虑的位置的5'方向上5个碱基为鸟嘌呤(g)和/或胞嘧啶(c)的百分比、从考虑的位置的5'方向上5个碱基的最长均聚物延伸和/或其组合。在一个实施方案中,上述段落中公开的所有模型特征、协变量和/或分类器都包含在变异识别模型中。在优选的实施方案中,上述段落中公开的所有模型特征、协变量和/或分类器都包括在surascore和/或surascorebb变异识别模型中,并且该模型使用拷贝调整的评分来调整一个或更多个模型特征、协变量和/或分类器的评分。还预期了实施方案的变化。g.序列变异预期实施方案可以包括预测、识别等任何序列变异。序列变异的非限制性实例可以包括:单核苷酸多态性(snp);单核苷酸变异(snv);复杂的碱基变化,例如多核苷酸取代;结构变异、基因组拷贝数变化和重排、定量拷贝数估计和/或其组合。还预期本发明的序列变异可以与任何疾病、病状、状态、药物基因组应答(例如耐药性、敏感度和/或毒性)、其倾向性和/或其组合相关。非限制性实例可以包括癌症、糖尿病、肥胖症、感染、自身免疫性疾病、衰老、肾脏疾病、代谢综合征、神经病理学、脑血管疾病、阿尔茨海默病、心血管疾病、卒中、对药物敏感、对化合物敏感、对复合物敏感、药物毒性、化合物毒性、复合物毒性、耐药性、耐化合物性、耐复合物性和/或其组合。预期可以平行地或依次地分析多种变异。在某些实施方案中,分析的基因座或变异的数目可以至少或至多为2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、170、171、172、173、174、175、176、177、178、179、180、181、182、183、184、185、186、187、188、189、190、191、192、193、194、195、196、197、198、199、200、201、202、203、204、205、206、207、208、209、210、211、212、213、214、215、216、217、218、219、220、221、222、223、224、225、226、227、228、229、230、231、232、233、234、235、236、237、238、239、240、241、242、243、244、245、246、247、248、249、250、251、252、253、254、255、256、257、258、259、260、261、262、263、264、265、266、267、268、269、270、271、272、273、274、275、276、277、278、279、280、281、282、283、284、285、286、287、288、289、290、291、292、293、294、295、296、297、298、299、300、301、302、303、304、305、306、307、308、309、310、311、312、313、314、315、316、317、318、319、320、321、322、323、324、325、326、327、328、329、330、331、332、333、334、335、336、337、338、339、340、341、342、343、344、345、346、347、348、349、350、351、352、353、354、355、356、357、358、359、360、361、362、363、364、365、366、367、368、369、370、371、372、373、374、375、376、377、378、379、380、381、382、383、384、385、386、387、388、389、390、391、392、393、394、395、396、397、398、399、400、401、402、403、404、405、406、407、408、409、410、411、412、413、414、415、416、417、418、419、420、421、422、423、424、425、426、427、428、429、430、431、432、433、434、435、436、437、438、439、440、441、442、443、444、445、446、447、448、449、450、451、452、453、454、455、456、457、458、459、460、461、462、463、464、465、466、467、468、469、470、471、472、473、474、475、476、477、478、479、480、481、482、483、484、485、486、487、488、489、490、491、492、493、494、495、496、497、498、499、500、501、502、503、504、505、506、507、508、509、510、511、512、513、514、515、516、517、518、519、520、521、522、523、524、525、526、527、528、529、530、531、532、533、534、535、536、537、538、539、540、541、542、543、544、545、546、547、548、549、550、551、552、553、554、555、556、557、558、559、560、561、562、563、564、565、566、567、568、569、570、571、572、600、700、800、900、1000个基因座或变异,或其中可得出的任意范围。h.比对序列预期本发明的实施方案可以包括将序列数据与一个或更多个参考序列比对。参考序列的非限制性实例包括:生物序列、非生物序列、合成序列、植物序列、动物序列、真菌序列、原核生物序列、人序列、正常组织序列、癌组织序列、患病组织序列、先前序列、来自遗传相关的生物体或家族成员的序列、基于种群的一般或特定遗传学的序列、人工序列、来自标准品的序列、来自文库中另一个样品的序列、来自相同样品的序列和/或其组合。i.方法预期本发明的实施方案可以包括方法和过程。方法的非限制性实例包括用于训练变异识别模型的方法、用于将可行模板计数并入到变异识别模型中作为模型特征的方法、用于将基于pcr的富集工作流程的元素与样品鉴定和生物信息学整合的方法。将基于pcr富集工作流程的元素与样品鉴定和生物信息学整合的方法的非限制性实例包括:包括样品鉴定的方法、pcr富集、标记pcr、纯化、文库定量、仪器加载、数据分析和报告(图1);包括定量和/或抑制剂分析的方法,例如qc分析;基因特异性pcr;标记pcr;纯化和大小选择;文库定量;归一化和混池、稀释和加载;测序,例如通过使用miseq;数据分析、变异识别和报告,例如通过使用reporterbioinformatics(图2a和2b以及图3a和3b)。j.试剂盒还预期试剂盒用于本发明的一些方面。例如,本发明的装置可以包括在试剂盒内。试剂盒可以包括一个或更多个容器。容器可以包括瓶子、金属管、层压管、塑料管、分配器、压力容器、阻隔容器、包装、分室、或其中保存了装置或所期望的瓶子、分配器或包装的其他类型的容器,例如注塑成型或吹塑成型的塑料容器。试剂盒和/或容器在其表面上可以包括标记。举例来说,标记可以是单词、短语、缩写、图片或符号。试剂盒还包括:一种或更多种定量pcr试剂;一种或更多种多重pcr试剂;一种或更多种标记pcr试剂;用于纯化和/或归一化来自样品或扩增目标的核酸的一种或更多种试剂;包括指令的一种或更多种计算机可读存储介质,所述指令当被处理器执行时引起处理器完成用于从测序数据文件中识别序列变异的方法;提供访问一个或更多个本地或远程计算机可读存储介质的一个或更多个指令,所述指令包括当被处理器执行时引起处理器完成用于从测序数据文件中识别序列变异的方法;一种或更多种引物,一种或更多种探针,一种或更多种标准品,一种或更多种阳性和/或阴性对照,一种或更多种合成批次对照;一种或更多种缓冲剂;一种或更多种稀释剂;和/或一种或更多种聚合酶或其它核酸修饰酶。试剂盒还可以包括使用试剂盒组件的说明书,在试剂盒中包含的任何其他产品的用途,或在试剂盒中不包含的其他产品的用途,例如但不限于软件或基于网络的应用。说明书可以包括如何应用、组装、使用和维护产品和/或组件的说明。在一个实例中,试剂盒可以提供用于将基于pcr的富集工作流程的元素与样品鉴定和生物信息学整合的组件或说明书。在另一种情况下,试剂盒可以遵循以下工作流程:样品鉴定、pcr富集、标记pcr、纯化、文库定量、仪器加载、数据分析和报告(图1)。在又一个实例中,试剂盒可以包括针对定量和/或抑制剂分析的组件,例如dnaqc分析;基因特异的pcr;标签pcr;纯化和大小选择;文库定量;归一化和混池、稀释和加载;测序,例如通过使用miseq;数据分析、变异识别和报告,例如通过使用reporterbioinformatics(图2a和2b以及图3a和3b)。在一个方面,试剂盒可以使由人组织或细胞系纯化的核酸的多个基因中的癌症相关变异定量和富集。在另一个方面,试剂盒含有或支持以下中的一种或更多种:支持使用特定仪器如illuminamiseq仪器进行多重下一代测序分析;包括分析测序数据文件例如miseq数据文件的软件,用于识别碱基取代突变和小插入/缺失;使用本地整合的生物信息管线;和/或使用伴随数据的可视化工具。在另一个方面,试剂盒可以包括包含例如引物、探针、rox和标准品的一种或更多种dna分析试剂盒;核心试剂如pancancer引物、ffpe阳性对照、合成批次对照、taq、缓冲剂母料混合物、稀释剂;包含例如珠、洗脱缓冲剂、洗涤缓冲剂的beadpurification;(miseq)组件,其包含例如母料混合物、rox、稀释剂、引物/探针、标准品、阳性对照和校准工具;miseqindexcodes引物混合;标签试剂和定制miseq引物组件,其包含例如母料混合物、稀释剂和定制测序引物(图4)。在又一个方面,试剂盒可以包含或进一步包含安装程序,用于安装本地应用的网络或现场展开的数据分析包(图4)。在另一个实例中,试剂盒可以包括用于确定可行模板计数和/或抑制曲线的组件。在具体的实施方案中,这种组件是ngs试剂盒。ngs试剂盒可以含有以下试剂中的一种或更多种:试剂结合在最小的瓶中用于简单设置和工作流程的2×母料混合物,易于使用和重复使用的预稀释标准品,和/或用于仪器相容性的ro校正染料(passivedye)(图4)。在另一个实例中,用于确定可行模板计数和/或抑制曲线的组件确定qfi分析评分和抑制(cq)(图5)。在一个方面,试剂盒可以包括基因特异性和标记pcr。试剂盒可以使用使用2步骤的pcr用于基因特异性和标记pcr的工作流程。在另一个方面,pcr的2个步骤可以是:(i)利用连接到每个引物的共同序列的基因特异性扩增;和(ii)第二pcr附加仪器-特异性连接头和索引编码被添加到pcr产物。在又一个方面,试剂盒还可以包括其中将来自各样品的产物混池,然后聚集到一个或更多个流动池上,在成像后,使用索引编码去卷积每个样品的每个扩增子的同一性(图6a和6b)。在一个实例中,试剂盒的基因特异性和标记pcr组件包括至少一种基因特异性的母料混合物和标签母料混合物。在另一个实例中,至少一种基因特定性的母料混合物和标签母料混合物包括以下:母料混合物设置-92个引物对的引物混合(3545-1)、与ngs试剂相同的2×pcr母料混合物(3469-1)、固定体积为4μl的样品;和/或用于标记pcr-寡核苷酸作为预混合物的“无母料混合物”设置、2×母料混合物(3469-1)和基因特异性产物的等分试样(图7)。在另一个方面,试剂盒可以包括目标面板和/或阳性对照。在一个实例中,试剂盒包括残留的临床ffpe来源的dna对照。在另一个实例中,过程对照由与基因组dna混合的并代表几种不同变异的几种合成dna配制。在又一个实例中,试剂盒对照代表癌症相关变异。在一个实例中,试剂盒对照由brafv600e阳性和“野生型”肿瘤配制。在又一个方面,试剂盒可以包括文库纯化、定量和加载组件。在一个实例中,文库纯化从多重pcr中除去游离的pcr引物和缓冲剂组分和/或减少非特异性引物二聚体产物。在另一个实例中,在样品加载前使用文库定量作为内部质量控制检查和/或在混池前使样品文库之间的产率归一化。在又一个实例中,通过珠纯化进行文库纯化。珠纯化的非限制性实例包括基于磁珠的纯化。在一个实例中,文库定量方法是无校准曲线的qpcr方法。定量方法的非限制性实例包括具有用于浓度确定的标样标准的竞争性pcr,其使用δct来确定每个文库的浓度。在另一个实例中,将加载组件与测序引物预先混合至指定浓度并随试剂盒一起提供。在又一个实例中,对于加载组件,用户将样品混池,使用phix变性,稀释并加载到盒中。在加载组件的一个实例中,用户提供双索引编码列表,并将结果与用于分析的fastq文件连接。在一个方面,试剂盒可以包括生物信息学组件。在一个实例中,生物信息学组件是用训练数据组研发的。在另一个实例中,将生物信息学软件提供以使用户能够分析产生的原始ngs数据,例如通过suraseq或pancancerdna面板产生的。在又一个实例中,软件将是安装在用户本地机器上的独立工具。在一个实例中,软件能够通过网页浏览器上下文中呈现的图形界面来使用。在另一个实例中,不需要互联网连接以使用软件。在又一个实例中,网页应用将从以无头模式运行的虚拟机托管,作为其安装到的机器上的窗口服务,并且可以由本地网络上的任何其他机器访问。在一个实例中,软件将服从hipaa和/或满足访问控制、审核控制、完整性、认证和传输安全的技术保障。在另一个实例中,软件将使用户能够通过点击式界面从如pgm或miseq仪器的测序仪器上加载原始序列数据,上传ngs数据,并启动产生样品质量控制和/或检测到的突变和评估检测到的变异的功能后果的信息的简明总结的分析。在另一个实例中,软件将支持导出结果或长期存储。在又一个实例中,生物信息学分析被跟踪并通过项目仪表板提供给用户。在一个实例中,所有的生物信息学处理都在运行windows主机环境的linux虚拟机上进行。在另一个实例中,生物信息学分析在特定的一组核酸序列上训练和/或提供变异性(参见图8a和8b作为非限制性实例)。在又一个实例中,变异识别器仅在400拷贝输入处识别真正的变异(参见图9作为非限制性示例)。实施例包括以下实施例以证明本发明的优选实施方案。本领域技术人员应理解以下实施例中所公开的技术能代表本发明人发现的在本发明的实践中发挥良好作用的技术,因此能够认为其构成用于其实践的优选实施方案。然而,根据本公开,本领域技术人员应理解在不脱离本发明的精神和范围的情况下,在所公开的具体实施方案中可以做出许多变化,并仍获得相同或相似的结果。实施例1实施和不实施可行模板计数特异性特征的变异识别模型的比较为了评估可行模板计数和可行模板计数相关的特征对变异识别器性能的影响,发明人训练了基线模型,其包括除了那些是可行模板计数特异性的特征的所有特征,和包括基线特征加可行模板计数特异性特征的可行模板计数模型(“可行的识别器”)。使用dna分析确定可行模板计数(改编自sah等人,2013)。具体地,使用以下记录的参数和特征训练模型。工作流程如图3a和b所示。材料和方法dna制备和测序通过dna分析评估dna功能性(改编自sah等人,2013)。dna分析指导了ngs富集步骤中的输入以确保变异识别的准确性。参见图3a和b。使用ngs试剂进行基于pcr的目标富集(从hadd等人,2013改进)。根据制造商的说明书,遵循miseq(illumina)和pcm(thermofisher)的测序程序。利用通过液珠阵列(luminex)(333)和/或复制体测序(467)的验证测序,并考虑位点和样品特异性背景后考虑一致识别阳性来确定突变状态。测序分析通过asuragen的标准预处理管线进行测序分析,其包括:扩增子相似性过滤(基于使用bfast比对器的带状smith-waterman与目标扩增子组的比对;连接头和pcr引物修剪;长度过滤(去除短于20个核苷酸的读取结果);边缘质量修剪(从扩增子边缘修剪低质量碱基(<q20);质量评分过滤(保留平均质量评分>20的读取结果);n过滤(排除其中具有n的读取结果);使用bwa与grch37比对(sw算法);使用来自1000个基因组、dbsnp和cosmic的已知插入和缺失和snv,gatk插入缺失-重新比对和碱基q评分重新校准(用于插入和缺失标记重新比对)。根据推荐的方案(koboldt等人,2013),使用varscan2进行变异识别(koboldt等人,2012)。模型参数及特征训练模型并经过5次交叉验证来评估性能。报告的性能是训练中使用的位置的平均交叉验证评分,以及在训练期间未使用的位置的模型预测评分(参见以下训练中使用的数据集)。使用以下参数,使用r(版本3.0.2)中的“ada”程序包(版本2.0-3)实施ada提升树:迭代:250促进收缩参数(boostingshrinkageparameter)“nu”:0.05样品取样包的取样率:1(即无随机取样)树深:5类型:真实所有其他参数都保留为默认值。通过两个评分指标(surascore和surascorebb)、制成表的数据和通过asuragen编写的自定义脚本添加的序列上下文指标评分最终的bams。该数据集代表超过1280个测序的样品,其由474个独特的样品组成(一些样品被测序多于两次)。通过以下方式挑选训练数据集:除去观察到的百分比变异小于0.5%的假设。(留下~250000个假设);从250k可用中选择50000个假设的随机组;将随机集与所有推定的体细胞变异和150个随机选择的推定的种系变异结合起来,总计约52000个假设。为了确保在相同数据集上训练基线模型和可行的模型,随机数生成器种子在随机选择前手动地设置为已知种子,提供数据的一致随机子集。训练数据集累积了474个独特的样品集,其包括:8个癌细胞系混合物、2个hapmap样品(na12878和na19240)、由等位基因频率范围为1%至40%突变体的基因组dna的背景中的46个gblock突变(可以通过万维网在idt.com/的取得)构成的2个合成对照,、18种血浆样品、171种临床ffpe、254种细针抽吸(fna)和19种新鲜冷冻样品。使用以下目标扩增子测序面板中的一种或更多种对这些样品测序:tp53面板,其涵盖了典型tp53的所有编码外显子;suraseq500;informagen+,一种由68个总扩增子组成的两池面板;suraseq200;pancancer面板,具有46个总扩增子的、单管形式的suraseq500面板延展。总的来说,测序的内容代表超过6kb的人基因组,富集了已知在各种癌症中具有高临床相关性的热点区域。选择的样品是至少两次重复测序的,和/或通过一些其他突变检测方法,包括luminex和数字pcr需要检测的那些样品。如果可行和通过重复一致,则通过与另外的检测方法对比确立真实性。特别地,跨越复制样品中的所有复制位点,基于观察到的百分比变异的最低95百分位,以位置特异性方式建立平均值和标准偏差的简单模型,如果观察到的百分比变异高于跨越所有重复的平均值+2标准偏差,则识别候选的突变。通过样品特异性假设标准进一步精选候选突变,其中观察到的突变必须大于讨论中的样品所观察到的假设特异性背景的第95百分位的2倍。上述唯一的例外是brafv600e,其中包含了发明人集合中的阳性富集代表,因此需要较低的位置特异性截止以识别如通过另外的方法论确定的已知阳性变异。结果如图10a和10b所示,具有低可扩增拷贝的样品将样品置于高假阳性和高假阴性率的风险中。这里的样品和设计用于训练具有和不具有包含具有低可行模板计数的样品的dnaqc分析数据(参见图11的策略概述)的分类器。具有或不具有dnaqc分析数据的变异识别器显示阳性变异数据分为了具有特征双峰等位基因频率分布的推定的种系变异和显示倾斜为较低丰度变异的推定的体细胞变异。参见图12。总之,数据表明体细胞变异与种系变异的合理近似。当与先前评估的方法相比时,基线模型和可行的模型在灵敏度方面均胜过竞争对手。图13显示了独立评估的其它方法的灵敏度,而图14显示了方法的可比较统计的灵敏度和ppv;注意到varscan是图13和图14中的共同元素,且注意到其可以实现可比较的灵敏度,并且在两个图中都具有相似的形状,注意到varscan显著地获得约20%变异的灵敏度。图15表明具有适当特征向量的机器学习方法可以实现关于等位基因频率的高灵敏度和特异性,其优于那些通过当前代的识别器实现的灵敏度和特异性,不考虑信息学包含。如图16所示的具有推定的种系变异的性能还显示了对于两种机器学习方法更好的灵敏度和ppv。然而,如图15所示,当考虑根据拷贝数的灵敏度和性能时,对于具有<100个功能性拷贝的样品,观察到相比于基线模型ppv(阳性预测值:真实变异的识别变异的百分比)约50%的提高。性能的这种提高可以直接地归因于将dnaqc分析拷贝数信息包含在模型中,因为所有其他变量、训练计划和训练参数都保持不变。100个拷贝数标记是高度相关的,因为在所评估的超过600个ffpe样品的队列中,超过27%具有每10ng(10ng是常见的分析输入格式)基因组dna输入少于100个拷贝(参见图17),其说明超过27%的样品将受益于通过显著减少假阳性数将qc数据直接并入变异识别模型中,甚至相对于市场上其他当前代的变异识别器相关的已经显著减少假阳性的模型。此外,可行的识别器显示了与低量、低质量的残留临床ffpedna一致的变异检测。将brafv600e阳性ffpe滴定到braf野生型ffpe样品的背景中至2.5%变异。功能性拷贝滴定为30至660。使用经训练的信息学模型识别样品。图10a和10b显示了变异识别的总数。点通过理论braf百分比着色,并且已经被抖动以避免过度绘制。图18显示观察到的变异等位基因频率与功能性拷贝输入。点通过理论braf百分比遮蔽,并根据braf识别的(三角形)或不识别的(圆圈)成形。识别器甚至在低复制输入和低百分比变异下保持高灵敏度和ppv。具体地,信息学模型识别残留临床ffpe中的braf变异,其中仅有34个和70个功能性拷贝输入,分别表示仅3.74(11%变体)和1.96(2.8%变异)突变体拷贝。结果表明,并入样品特异性实验信息改善了突变检测的灵敏度和特异性,特别是对于ffpe和fna活组织检查中的低流行变异。在低质量和低数量dna样品中识别变异的能力增加了可以以高置信度处理的临床样品数。发明人还证明了对于肿瘤标本和参考细胞系材料的确定混合物0.5%至10%流行率的变体,具有高灵敏度和ppv的变异识别。结果强调了实行可行模板计数的识别系统的价值。实施例2asuragenngspan-cancerdna面板为了评估包含试剂和分析工具的试剂盒的性能,该分析工具包括可行的识别器,研发了ngspan-cancerdna面板(图2b)并使用来自人组织或细胞系纯化的dna的21个基因中癌症相关的变异测试。工作流程和具体步骤和组件在图2a至图9中举例说明。试剂盒支持使用illuminamiseq仪器的多重下一代测序分析。试剂盒包括使用本地整合的生物信息学管线和伴随数据可视化工具分析miseq数据文件用于识别碱基替代突变和小插入/缺失的软件。具体地,试剂盒包括(1)包含引物、探针、rox和标准品的dnaqc分析试剂盒;(2)包含quantidexpancancer引物、ffpe阳性对照、合成批次对照、taq、缓冲剂母料混合物(matermix)、稀释剂的pancancer核心试剂组分;(3)quantidexpureprep珠纯化组分,其包括磁珠、洗脱缓冲剂和洗涤缓冲剂;(4)(miseq)组件,其包含2x母料混合物、rox、稀释剂、引物/探针、标准品、阳性对照和校准工具;(5)codesmiseq索引编码(1-24)引物混合;(6)标记试剂和定制的miseq引物组分,其包含2x母料混合物、稀释剂和定制的测序引物;(7)包括安装程序的数据管线、分析和报告工具组件以及用于作为本地应用安装的网页或现场部署的数据分析包(图4)。变异识别器是可行的识别器(reporter)。使用qpcr确定qfi分析评分和抑制曲线的试剂包括将2x母料混合物与试剂组合在最小的瓶中用于简单设置和工作流程、易于使用和重复的预稀释标准品,以及用于仪器相容性的rox校正染料。样品队列减轻如图5所示。asuragenngs工作流程使用两个步骤的pcr:(i)利用连接到每个引物的共同序列的基因特异性扩增;(ii)第二pcr附加仪器-特异性接头和索引编码添加到pcr产物中。将来自各个样品的产物混池,然后聚集到流动池上。成像后,索引编码用于对每个样品的每个扩增子的识别进行去卷积。将方案设计为用于简单处理和最小试剂。其包括(1)包含92个引物对的引物混合(3545-1)、与相同的2×pcr母料混合物(3469-1)、固定体积为4ml的样品;(2)用于标记包含寡核苷酸的pcr作为预混合物的“无母料混合物”设置、2×母料混合物(3469-1)和基因特异性产物的等分试样。试剂盒包括两个阳性对照、过程对照和ffpe阳性对照。过程对照由14种合成dna混合基因组dna配制,代表14种不同的癌症相关变异。ffpe阳性对照由brafv600e阳性和“野生型”肿瘤块配制。由发明人研究验证运行ms127的结果总结在表1中:表1操作者变异读取结果百分比1brafv600e5.32brafv600e3.93brafv600e6.5纯化文库使用基于磁珠的纯化,其使用以下过程:结合、洗涤、洗脱、设计为减少<190bp的产物并保留特定的产物。文库定量是使用用于浓度确定的加标标准的竞争性pcr的简单、无校准曲线的qpcr方法。方法在提供的标准拷贝数的100倍范围内工作。方法使用δct来确定每个文库的浓度。还可以使用其它文库定量方法,例如使用依赖于标准曲线确定文库中模板分子拷贝数的dna插入染料或qpcr分析。仪器加载使用与asuragen的定制seq引物预先混合至指定浓度的illumina的标准测序引物并随试剂盒一起提供。试剂盒设计为使用户混池样品、用phix变性、稀释并加载到盒中。然后用户提供双索引编码列表,并将dnaqc结果与fastq文件连接用于分析。生物信息学使用直观的生物信息学软件选项,其使用户能够分析由pancancerdna面板产生的原始ngs数据。研发了原型用户界面以支持由虚拟机托管的管线的点击操作,并重新使用surasight或reportergui组件使结果可视化。原型允许用户登录,创建分析项目,上传原始序列数据并启动分析。分析的状态被跟踪并通过项目仪表板提供给用户。一旦分析完成,可以从界面下载打包的surasight或报告。所有该处理都在运行windows主机环境的linux虚拟机上发生。已经研发了通过点击的安装程序,其证明了通过标准安装向导在主机上安装虚拟机的可行性。结果使用上述试剂盒测试总共90个总dna样品。试剂盒在5x中位数读取结果中产生100%扩增子的中值。在24个样品/运行的标度值下,ffpe样品中的扩增子都不具有<500读取结果的覆盖深度,ntc~4至6中值读取结果/扩增子。试剂盒在多操作臂中产生2至6%cv的ffpe突变定量。通过所有操作者检测5%brafffpe对照(3.9%、5.3%、6.5%)。在5%、8%、10%和12%的合成对照在变异丰度上是内部一致的。试剂盒提供了用已知插入和缺失和cnv的dna样品的成功检测。存在来自抑制的ffpedna的文库产物的剂量依赖。如图8a和b所示,扩增子的产量、总体覆盖率和操作者之间的变化性突出了面板的性能。此外,使用获知的变异识别,才能在400个拷贝输入中识别真正的变异,减少了分析的复杂性并确认或拒绝假阳性结果(图9)。实施例3每个功能性拷贝的asuragen变异识别器性能在多操作者、多天、多运行研究中总共98个样品被测序。评估在5%变异等位基因频率(var)或以上的变异的变异识别器性能,并通过功能性拷贝输入分离到文库中。在200个拷贝输入中,发明人观察到完美的性能,但在低于200个拷贝其与敏感性和阳性预测值(ppv)增加的风险相关。结果总结在表2中:表2功能性拷贝输入预期的变异数灵敏度ppv≤200310.870.93>20034011实施例4每个功能性拷贝的erbb2基因上的asuragen变异识别器性能用靶向erbb2基因的面板对51种不同质量的石蜡包埋(ffpe)样品测序。在可用测序读取结果(y轴)和输入到测序反应的功能性拷贝(x轴)中的百分比存在明确的关系,>1000个拷贝提供最好的结果,>200个拷贝提供足够的结果(图19)。拟合线:具有95%ci的loess平滑线。实施例5用于与ddpcr比较的cnv的asuragen变异识别器性能在erbb2基因座具有已知和变化的拷贝数变化(cnv)的实施例4的51个样品使用设计有cnv检测能力的erbb2靶向面板测序。通过微滴式数字pcr(ddpcr)(bioradsep25)对cnv定量评估相同的样品(图20)。数据显示两种方法之间的强相关性。实施例6基于样品质量的扩增子性能的asuragen变异识别器性能靶向扩增子面板中的cnv检测依赖于扩增子相对于彼此一致的扩增效率。然而,相对的扩增效率根据样品质量变化。显示的是使用实施例4的51个样品的样品间相对扩增效率的标准偏差。随着dna质量评分(qfi)降低,相对效率差异加剧,导致偏离预期基线增加(图21)。这证明扩增子性能取决于样品质量。实施例7asuragen变异识别器估计的功能性拷贝%与基于qpcr的方法比较通过qpcr测量样品的qfi的几种不同扩增子长度和损伤频率,并确定功能性%并与相同样品的ngs结果比较。用于估计样品损伤频率的基于ngs的方法,通过延伸,将用于任何大小范围的功能性dna%(brisco等人,2010)与用于测量相同信息的基于qpcr的方法充分比较(图22)。这表明预测序质量控制(qc)具有对相对扩增效率和通过延伸可靠地识别cnv的能力的直接影响。实施例8asuragen变异识别器与没有考虑输入拷贝数的识别器的比较低功能性拷贝增加qc不可知识别器中的假阳性识别(图10左格),但不增加braf(图10a)和kras(图10b)拷贝数滴定研究中的识别器(图10右格)。实施例9独特的外显子含量和四种潜在qc方法之间的相关性进行用于独特外显子含量的四个潜在质量控制方法的比较,独特的外显子含量通过整个转录组rna-seq确定。比较以下qc方法:生物分析仪(dv200:大于200个核苷酸的片段%)、纳米滴(质量)、量子位rna(质量)和quantidexrnaqc(功能性拷贝)。对于每个qc方法评估适合于独特外显子读取结果的r2值。结果证明rnaqc(测量功能性rna拷贝的基于rt-qpcr的分析)提供了比其他方法更准确的结果。结果汇总于表3中。表3这些结果还证明使用rna功能性拷贝评估的rnaqc比另外的qc方法更能预测整个转录组数据质量和测序质量。实施例10rna功能性拷贝试验的分析可以用于拯救较低质量的样品,并提供读取结果准确性的更好预测可以通过增加文库质量输入(图23)来拯救较低质量的ffpe样品(通过rnaqc确定的rna功能性拷贝分析来分级)。rna功能性拷贝数也预测了测序数据质量。通过rnaqc确定,具有每2ulrt的内源性对照rna的小于100个rna功能性拷贝的文库显示出显著减少的预期目标的比对率(图24)。rna功能性拷贝数评估也是假阴性融合识别风险的预测。使用两个融合基因ret/ptc1和pax8-pparg的dna样品和阴性对照(bwh-107a)来确定在不接受假阴性可以使用的平均功能性rna拷贝定义的最小量的样品。结果汇总于表4中。表4根据由ngs产生的目标上的读取结果绘制通过rnaqc确定的rna功能性拷贝。图显示了rna功能性拷贝与目标读取结果之间的高相关性(图25)。输入质量似乎没有像所测试的样品的相似输入质量的扩散所证明的那样高。这证明在测序前使用rna功能性拷贝分析来修改每个样品的样品量/功能性拷贝数可以提高所产生的测序数据的质量。这也证明在识别方法中考虑rna功能性拷贝可以更好地帮助确定读取结果的准确性。此外,这证明rna功能性拷贝相比所使用的样品质量是读取结果准确性的更好预测。本文中所公开和要求保护的所有装置和/或方法可以根据本公开不需要过度实验完成和实现。尽管本发明的装置和方法已经按照优选的实施方案进行了描述,但是对于本领域技术人员明显的是,可以对所述装置和/或方法以及在本文所描述方法的步骤或步骤的顺序中实施变化,而不脱离本发明的概念、精神和范围。对本领域技术人员明显的相似替代和改变都视为在如由所附权利要求限定的本发明的精神、范围和概念内。参考文献以下参考文献在一定程度上提供示例性程序或对本文所陈述细节的其他补充细节,通过引用明确地并入本文。美国公开号2012/0322058美国公开号2014/0057793美国公开号2014/0058681ep2602734a1wo公开号2013/159145akbarim,hansenmd,halgunsetj,skorpenf,krokanhe:lowcopynumberdnatemplatecanrenderpolymerasechainreactionerrorproneinasequence-dependentmanner.jmoldiagn2005,7:36-39.beltranh,yelenskyr,framptongm,parkk,downingsr,macdonaldty,jaroszm,lipsond,tagawast,nanusdm,stephenspj,mosquerajm,croninmt,rubinma:targetednext-generationsequencingofadvancedprostatecanceridentifiespotentialtherapeutictargetsanddiseaseheterogeneity.eururol2013,63:920-926.briscomj,morelyaa:quantificationofrnaintegrityanditsuseformeasurementoftranscriptionnumber.nucleicacidsres2012,40(18):e144.briscomj,lathams,bartleypa,morleya.:incorporationofmeasurementofdnaintegrityintoqpcrassays.biotechniques201049:893-897.didelota,kotsopoulossk,lupoa,pekind,lix,atochini,srinivasanp,zhongq,olsonj,linkdr,laurent-puigp,blonsh,hutchisonjb,talyv:multiplexpicoliter-dropletdigitalpcrforquantitativeassessmentofdnaintegrityinclinicalsamples.clinchem2013,59:815-823.forshewt,murtazam,parkinsonc等人:noninvasiveidentificationandmonitoringofcancermutationsbytargeteddeepsequencingofplasmadna.sci.transl.med.2012,4(136):136ra1681.gargisas,kalmanl,berrymw,bickdp,dimmockdp,hambucht,luf,lyone,voelkerdingkv,zehnbauerba等人:assuringthequalityofnext-generationsequencinginclinicallaboratorypractice.natbiotechnol2012,30:1033-1036.haddag,houghtonj,choudharya,sahs,chenl,markoac,sanfordt,buddavarapuk,krostingj,garmirel,wylied,shinder,beaudenons,alexanderek,mamboe,adaiat,lathamgj:targeted,high-depth,next-generationsequencingofcancergenesinformalin-fixed,paraffin-embeddedandfine-needleaspirationtumorspecimens.jmoldiagn2013,15:234-247.koboldtdc,zhangq,larsonde,shend,mclellanmd,linl,millerca,mardiser,dingl,wilsonr:varscan2:somaticmutationandcopynumberalterationdiscoveryincancerbyexomesequencing.genomeres2012,22(3):568-576.menonr,dengm,boehmd,braunm,fendf,boehmd,biskups,perners:exomeenrichmentandsolidsequencingofformalinfixedparaffinembedded(ffpe)prostatecancertissue.intjmolsci2012,13:8933-8942.sahs,chenl,houghtonj,kemppainenj,markoa,zeiglerr,lathamg:functionaldnaquantificationguidesaccuratenext-generationsequencingmutationdetectioninformalin-fixed,paraffin-embeddedtumorbiopsies.genomemedicine2013,5:77.sedlackovat,repiskag,celecp,szemest,minarikg:fragmentationofdnaaffectstheaccuracyofthednaquantitationbythecommonlyusedmethods.biolprocedonline2013,15:5.simbolom,gottardim,corbov,fassanm,mafficinia,malpelig,lawlorrt,scarpaa:dnaqualificationworkflowfornextgenerationsequencingofhistopathologicalsamples.plosone2013,8:e62692.tuononenk,-nevalas,sarhadivk,wirtanena,m,salmenkivik,andrewsjm,telaranta-keerieai,hannulas,s,ellonenp,knuuttilaa,knuutilas:comparisonoftargetednext-generationsequencing(ngs)andreal-timepcrinthedetectionofegfr,kras,andbrafmutationsonformalin-fixed,paraffin-embeddedtumormaterialofnon-smallcelllungcarcinoma-superiorityofngs.geneschromosomescancer2013,52:503-511.vanbeerseh,joossesa,ligtenbergmj,flesr,hogervorstfb,verhoefs,nederlofpm:amultiplexpcrpredictorforacghsuccessofffpesamples.brjcancer2006,94:333-337.wangf,wangl,briggsc,sicinskae,gastonsm,mamonh,kulkemh,zamponir,lodam,mahere,oginos,fuchscs,lij,haderc,makrigiorgosgm:dnadegradationtestpredictssuccessinwhole-genomeamplificationfromdiverseclinicalsamples.jmoldiagn2007,9:441-451.yostse,smithen,schwabrb等人:identificationofhigh-confidencesomaticmutationsinwholegenomesequenceofformalin-fixedbreastcancerspecimens.nucleicacidsres2012,40(14):e107.当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1