在配对的颊粘膜拭子和尿液样本中使用微卫星分析进行膀胱癌检测的制作方法
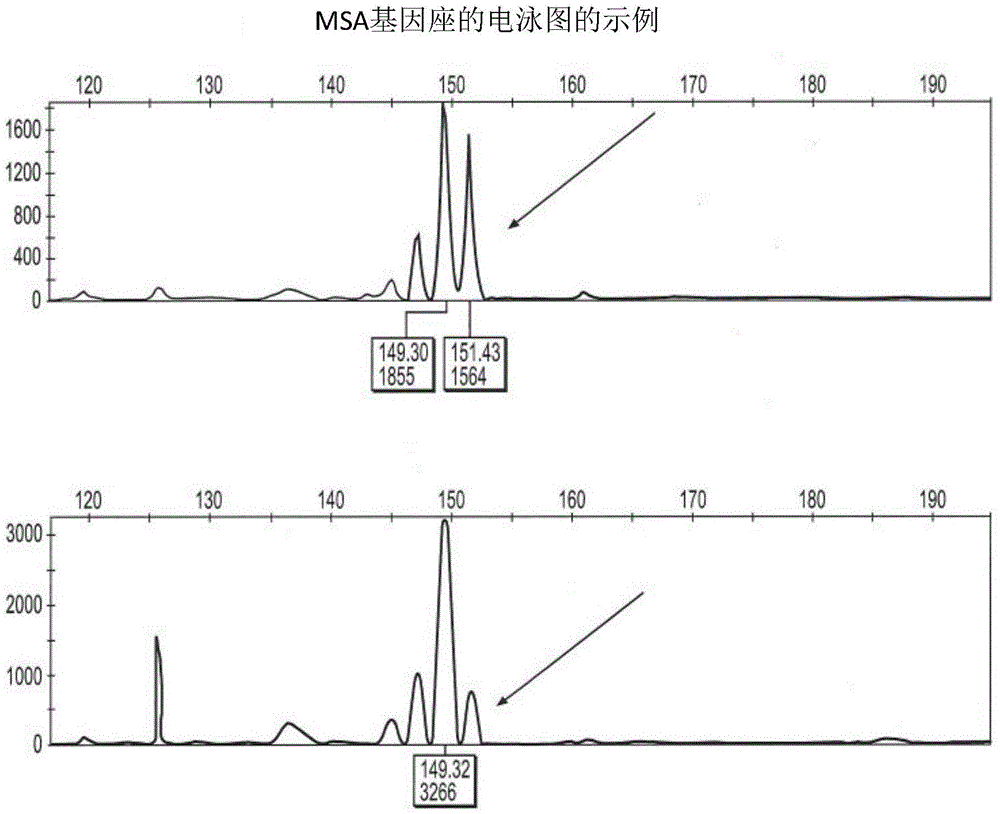
本申请基于2015年12月8日提交的美国临时专利申请号62/264,504的公开内容,并且要求上述申请提交日期的优先权,其全部公开内容在此通过引用并入本申请。发明领域本申请公开了可用于检测膀胱癌的组合物和方法。发明背景膀胱癌在美国男性和女性中分别是第4常见和第7常见的恶性肿瘤。在美国男性和女性中,每年分别诊断出60,000和18,000起膀胱癌的新病例。大约75%的患者患有该疾病的浅表型,并且这些患者的15%处于该疾病发展成侵袭型的风险。大约70%患有该疾病的浅表型的患者在10年内经历疾病复发,其中大多数的复发在初始诊断后的两年内检测到。由于这些原因,患有该疾病的浅表型的患者需要对疾病发展和复发进行监测。目前,膀胱癌监控的护理标准包括持续两年、每三个月一次的膀胱镜检查和尿细胞学检查,随后每年一次膀胱镜检查、尿细胞学检查和上尿路的x射线评估。然而,由于细胞学检查的灵敏度和特异性分别为仅25-50%和90-100%,这种监控方法并不是最优的。尽管膀胱镜检查的灵敏度较高(90-100%),这一程序收费昂贵,需要复杂的仪器以及无菌环境。而且,由于其侵入性的性质,这一程序具有固有的损伤、并发症和感染的风险。因此,需要既灵敏又对膀胱癌具有特异性的,并且与其他检测方法相比具有较低的并发症的风险的检测膀胱癌的方法。本申请公开的方法和试剂盒涉及上述的以及其他的重要需求。发明简述特别地,为了应对上述未满足的需求,本申请公开了在受试者中检测杂合性丢失的方法,包括:从膀胱样本和匹配的对照样本中扩增微卫星标记物的组以产生扩增产物的组,其中所述标记物的组包括fga、d9s747、mbp、d9s162、tho1、ifn-a、d21s1245和d20s48;检测所述扩增产物;比较来自所述膀胱样本的扩增产物和来自所述匹配的对照样本的扩增产物;和在所述膀胱样本中确定杂合性丢失是否存在于任意一个所述标记物中。还提供了在受试者中检测膀胱癌的方法,包括:使用本申请所述的引物从膀胱样本和对照样本中扩增微卫星标记物的组;检测所述反应产物;和在所述膀胱样本中确定是否多于一个的所述微卫星标记物展现出杂合性丢失或微卫星不稳定性,其中在至少两个微卫星标记物中展现杂合性丢失或不稳定性指示患有膀胱癌。本申请还公开了试剂盒,其包括具有特定核酸序列的引物以及所述引物的包装。附图简述结合附图阅读时,将进一步地理解该简述以及以下详述。为了说明公开的方法和试剂盒的目的,附图中示出了所述方法和试剂盒的示例性的实施方式;然而,所述方法和试剂盒不限于公开的具体实施方式。在附图中:图1显示了ifn-a基因座处的loh;图2显示了对d9s171基因座中另外的等位基因的检测;图3显示了在d16s476基因座中检测到的人工信号;图4提供了显示在str标记物处的loh的电泳图,所述str标记物位于9q32处;图5提供了用于研究患有膀胱癌的受试者的特征和相关样品号的汇总;图6a示出了对照受试者n1的msa数据;图6b示出了对照受试者n2的msa数据;图6c示出了对照受试者n3的msa数据;图6d示出了对照受试者n4的msa数据;图6e示出了对照受试者n5的msa数据;图6f示出了膀胱癌受试者c1的msa数据;图6g示出了膀胱癌受试者c2的msa数据;图6h示出了膀胱癌受试者c3的msa数据;图6i示出了膀胱癌受试者c4的msa数据;以及图6j示出了膀胱癌受试者c5的msa数据。发明详述通过参照以下详细描述,结合附图(其构成本公开内容的一部分),可以更加容易地理解公开的方法和试剂盒。应当理解,公开的方法和试剂盒不限于本申请描述和/或示出的具体的方法和试剂盒,并且本申请使用的术语是用于仅通过示例的方式描述特定实施方式的目的,而且不旨在对要求保护的方法和试剂盒进行限制。除非另外具体规定,否则关于可能的机制或作用模式或改进的理由的任何描述都仅仅是作为说明性的,并且公开的方法和试剂盒不会受到该建议的机制或作用模式或改进的理由的准确或错误的束缚。本说明书通篇涉及组合物和使用所述组合物的方法。当本公开描述或要求保护与组合物相关的特征或实施方式时,该特征或实施方式同样适用于使用所述组合物的方法。相似地,当本公开描述或要求保护与使用组合物的方法相关的特征或实施方式时,该特征或实施方式同样适用于所述组合物。在表达数值的范围时,另一实施方式包括从一个特定数值和/或至另一特定数值。另外,对以范围表述的数值的指代包括该范围内的每个数值。所有的范围都是包含性的以及可组合的。当以近似值表达数值时,通过使用前缀“约”,应当理解该特定数值构成另一实施方式。除非上下文另有明确规定,否则对特定数值的指代包括至少该特定数值。应当理解,在本申请中,为了明确起见而在不同的实施方式的上下文中描述的公开的方法和试剂盒的某些特征,也可以以组合的形式在单一实施方式中提供。相反地,为了简洁起见而在单一实施方式的上下文中描述的公开的方法和试剂盒的多个特征,也可以分别提供或以任何亚组合的形式提供。用于本申请中的单数形式“一个(a)”、“一种(an)”和“所述”包括复数形式。说明书和权利要求通篇使用多个涉及说明书各个方面的术语。除非另外指出,否则该术语应被赋予其在本领域中的常规含义。其他具体定义的术语应当以与本申请提供的定义一致的方式理解。用于本申请中的“微卫星”(与“简单序列重复(ssr)”和“短串联重复(str)”可互换地使用)是指核酸片段,其具有由2至6个碱基对组成的碱基单位的连续重复。微卫星通常长度为100-400个碱基对。因此,如果微卫星的碱基单位是4个碱基对,则会有约25至100个碱基单位的连续重复。“微卫星的不稳定性”是指当错配修复(mmr)没有正常运作时,细胞中微卫星的核酸序列发生的变化。由于丧失了dna聚合酶固有的纠正错配的校对能力,微卫星特有的重复单位容易出错。核酸碱基的插入或缺失的累积可导致框移突变,其对蛋白表达可造成有害的影响。聚合酶未检测到的错配错误由mmr纠正,因此mmr系统可以被视为一种失效保护系统。编码mmr系统的基因中的突变导致对微卫星复制缺乏监控,并且可导致不稳定性。已经将肿瘤抑制基因中发生的微卫星不稳定性与癌症表型相关联。术语“杂合性丢失”或loh是指丢失一段dna,其最有可能是一条染色体链而非另一条中的复制错误导致的。在一些癌症中,如果丢失的遗传材料编码全部或部分的肿瘤抑制蛋白、细胞繁殖机构或者细胞的稳定繁殖所必需的其他蛋白,则loh可能是特别成问题的。尽管余下的拷贝可能足以防止由丢失的等位基因所造成的任何有害影响,但如果余下的拷贝不足以防止有害影响,或者如果余下的拷贝被诱变,使得编码的蛋白无法起到野生型蛋白的作用,则loh可导致或至少促成癌症表型。余下的微卫星等位基因仍然易于突变,特别是在缺乏功能完整的mmr系统的细胞中更是如此,其可能得到进一步的产生无功能的蛋白的突变或变异,因此微卫星等位基因处的loh可能与癌症相关。如果所述无功能的蛋白是肿瘤抑制物,则癌症表型的风险增加。除每年诊断出的大约80,000起膀胱癌的新病例之外,每年大约有12,000位男性和4,500位女性死于该疾病。由于该疾病在美国国内外的盛行,进行了大量研究以识别可能促成该疾病或者另外指示患有该疾病的遗传组分。本研究识别了多种与该疾病在某种程序上相关联的肿瘤标记物,并且在过去的十年,已将几种肿瘤标记物纳入用于膀胱肿瘤检测的体外诊断测试。目前,btatm、btatmstat、fdptm和nmp-22tm已被食品药品监督管理局(fda)批准为膀胱学检查和细胞学检查的辅助物,并且这些测试通常是抗体介导的对肿瘤标记物的检测。这些测试的灵敏度和选择性数据表明,尽管这些标记物检测方法可能增强同时期的侵入性的检测程序,但是其灵敏度和选择性数据表明,其在没有结合诊断时则是不可靠的(halachmi等,molecularurology,1(4):309-314(1997);han和schoenberg,urologiconcology,5(3):87-92(2000))。例如,取决于疾病所处的阶段,blastat测试的灵敏度可以低至51%,并且对于1期癌症可以低至42%。相比之下,本发明是无需侵入性技术的检测膀胱癌的方法或者用于诊断的组合方法。更具体地说,本发明在膀胱细胞中通过分析来自膀胱样品的特定微卫星标记物的组来检测loh或msi,其中在膀胱样本中的两个标记物处检测到loh则指示患有疾病。在本发明的一些实施方式中,所述疾病是膀胱癌。尽管侵入性的技术对于获取测试样品不是必需的,但本发明不限于仅来源于非侵入性技术获取的样本。获取自活检、外科手术甚至尸检的样本可以用于使用在此描述的技术检测膀胱癌。在本发明的一些方面,膀胱样本可以是尿样,在其他方面,膀胱样本可以是膀胱活检样本。在本发明的一些实施方式中,匹配的对照样本是血液、血清、颊细胞、毛囊、唾液、皮脂、皮肤、汗液或泪液。在一些方面,匹配的对照样本是颊细胞。当微卫星与特定疾病相关联时,微卫星可以用作检测、诊断和/或研究疾病的标记物。本发明公开了检测微卫星标记物的组中的变化的方法。为了检测这些标记物,必须分离来自膀胱样品中的核酸。微卫星的扩增使本领域技术人员得以检测微卫星的存在、缺失或变化。微卫星两侧是可以用于设计用于扩增该微卫星的引物的序列。因此,在本发明的一些方面,使用一对引物扩增各标记物。表1记载了本发明的一个实施方式中用于扩增的各标记物的引物。使用任何能够扩增核酸的技术完成核酸的扩增,如等温扩增反应,其在基本等温的温度下扩增核酸,或者聚合酶链式反应(pcr),其使用不同温度的多个循环使模板核酸变性、使引物退火,以及延伸新形成的核酸链。pcr反应包括但不限于传统pcr、实时(rt)pcr和定性pcr(qpcr)。本发明涉及允许基于大小对扩增的核酸进行区别的任何扩增方案。在本发明的一个实施方式中,从获取自尿液样本和获取自颊样本的核酸扩增微卫星标记物。得到的扩增的核酸将具有预期的长度,和该预期的长度的任何衍生物,包括扩增产物的完全缺失,可能标示了微卫星不稳定性或杂合性丢失。本发明的一个实施方式提供了在受试者中检测loh的方法,包括从膀胱样本和配对的对照样本中扩增微卫星标记物以产生扩增产物,其中所述标记物包括fga、d9s747、mbp、d9s162、tho1、ifn-a、d21s1245和d20s48;检测所述扩增产物;比较来自所述膀胱样本的扩增产物和来自所述匹配的对照样本的扩增产物;和在所述膀胱样本中确定杂合性丢失是否存在于任意一个所述标记物中。可以使用本领域已知的分子方法分解扩增的核酸,包括但不限于琼脂糖凝胶电泳、毛细管电泳和高压液相色谱法。当比较来自膀胱样本的单一扩增产物和来自对比样本的单一扩增产物时,琼脂糖凝胶电泳允许目测样本是否具有标记物核酸。毛细管电泳还允许自动分解大小不同的扩增产物,并且这一方法尤其适合于在单一毛细管中分解多个扩增产物。本发明的一些方面提供了在单一扩增反应中扩增多个标记物。这种方法需要确定合适的反应条件(例如引物组合、离子强度、温度条件等)。由于一些标记物无法被有效地扩增,在扩增后无法被明确地识别,或者以有限的量存在,因此可能需要一个以上的扩增反应。因而,在一些实施方式中,标记物的扩增包括至少两个多重扩增反应。在一些方面,标记物的扩增包括三个多重扩增反应。在本发明的一些实施方式中,其中标记的扩增包括三个多重扩增反应,一个多重扩增反应中的待扩增的标记物包括fga和d9s747。在一些实施方式中,一个多重扩增反应中的待扩增的标记物包括d9162、mbp、ifn-a和tho1,或其任意亚组合。例如,一个多重扩增反应可以仅包括d9162和mbp。在一些实施方式中,一个多重扩增反应中的待扩增的标记物包括d21s1245和d20s48。尽管多重扩增反应可以是检测标记物的loh或msi的有效机制,但各标记物可以被单独扩增。随后在分析loh或msi之前可以将单独扩增的反应产物组合。各扩增产物也可以被单独分析,以确定loh或msi是否存在。表1列出了三个多重扩增反应(mp1、mp2和mp3)的特征,包括标记物名称;各标记物所位于的染色体(chr);各标记物的短串联重复序列;正向和反向引物;以及与正向引物缀合的染料。例如,标记物d4s243的str是(atag)n,其中“atag”是重复单元,且“n”是该单元重复的数量。d4s243位于4号染色体上,并且其正向引物用6-fam标记。表1在本发明中,可以标记扩增的核酸,以协助将一种扩增产物与另一种区分开。在本发明的一些方面,引物对中一个引物用荧光染料标记。表1指出了连接至正向引物的5’末端的荧光染料(“6fam”、“vic”、“ned”和“pet”)。染料的激发最大值不同(分别为520nm、554nm、575nm和595nm),其允许大小相似的扩增产物之间的区分。一旦连接至寡核苷酸,荧光染色常表现出其激发最大值的偏移;但任何的激发最大值将参照制造商记载的未缀合的染料的激发最大值。本发明还涉及能够连接至核酸引物的其他荧光染料。在一些方面,至少两个用于标记引物的荧光染料具有不同的最大荧光发射波长。当荧光标记的引物用于扩增微卫星标记物时,本发明的一个方面是使用能够检测发射荧光的遗传分析装置。在某些方面,遗传分析装置采用毛细管电泳来分离扩增产物和ccd照相机来检测来自产物的发射荧光。例如,abi3130遗传分析仪利用毛细管电泳(ce)按尺寸分离扩增产物。该装置还具有用于激发荧光染料的激光光源和用于检测荧光标记的扩增产物的ccd照相机。ce装置通常以电泳图的形式输出,其是电泳期间检测到的光的图形表现形式,因此允许观察者快速识别预期峰的存在,缺失或变化。图1描绘了ifn-a基因座的电泳图。顶部电泳图由源自颊粘膜拭子的标记扩增产物产生,而底部电泳图由源自尿液样本的标记扩增产物产生。颊样本和尿液样本来自相同的受试者,这允许确定膀胱样本中获得的loh或msi。例如,图1中的电泳图显示膀胱样本中的loh。颊样本中的最后一个主峰与前一个峰大小基本一致。这两个峰代表微卫星基因座的两个等位基因。在图1的膀胱样本电泳图中,仅有一个主峰,指示loh在该基因座处。还可采用通过使用定义的信号采集箱来识别str等位基因的软件。“箱(bin)”对应于预期检测扩增产物的特定区域。将图1中的箱表示为电泳图上方的细长矩形,且表明预期的扩增产物将在132至152碱基对范围内。ifn-a等位基因是箱中的主要峰。然而,对于一些微卫星,可以在指定的箱外检测到等位基因。例如,在图2的底部电泳图中的d9s171箱外部检测到d9s171微卫星中的纯合等位基因。图3说明了扩增产物中存在人工信号。该人工信号是d16s476基因座箱外的一个小峰。当分析样本的d16s476基因座中的msi或loh时,该人工信号不会被视为单独的等位基因。扩增反应的人工信号通常存在于膀胱和颊粘膜拭子样本的扩增产物中。如图3所示,人工信号在两个样本中都是恒定存在的。实施例提供以下实施例以进一步描述本发明所公开的一些实施方式。这些实施例旨在说明而非限制本发明所公开的实施方式。实施例1:颊粘膜拭子提取和基因组dna提取试剂制备和颊粘膜拭子处理a)制备步骤使用市售方案提取来自颊粘膜拭子的基因组dna。b)避免样本交叉污染和将拭子转移到微量离心管的方法为了减少或消除交叉样本污染的可能性,当添加洗涤试剂时仅打开一个管,如果手套与任何样本液体接触则更换手套。将颊粘膜拭子置于适当标记的1.5ml微量离心管中,并将盖子封闭在尖端的基部上,其中轴与尖端接触。将轴折断并丢弃到生物危害废物容器中,并关闭微量离心管。必要时用剪刀剪掉拭子轴,但在这种情况下,剪刀在样本之间用10%naclo(漂白剂)去污。c)从颊粘膜拭子中提取dna向每个样本管中加入400μl磷酸盐缓冲(pbs)溶液。将管剧烈涡旋50秒以确保拭子完全饱和。将管翻转10次以确保pbs接触管的所有区域。向每个样本管中加入20μlqiagen蛋白酶k储备溶液和400μl缓冲液al,并将管翻转75次。将样本充分涡旋50秒以确保适当的裂解并在56℃下孵育60分钟。将管短暂离心以除去冷凝液。向每个样本中加入400μl100%乙醇,将管倒置50次或1分钟,然后剧烈涡旋50秒。将管短暂离心以除去冷凝液。将700μl带有乙醇的裂解液加到相应的qiaamp旋转柱,不润湿边缘,且关闭盖子。避免用移液管尖端接触qiaamp膜。以8000rpm将离心管离心1分钟,弃去含有滤液的离心管。将qiaamp旋转柱置于干净的2ml收集管中。将尼龙拭子从1.5ml管中取出并置于适当标记的旋转柱中。当从棉签中提取dna时,将拭子垂直地擦拭到柱的边缘以使棉纤维从木材上松开,从而使拭子脱离木棒并留在柱中。将离心管以6000xg(8000rpm)离心1分钟,弃去含有滤液的离心管。向每个柱中加入500μl缓冲液aw1而不润湿边缘。盖上盖子,将离心管在室温下孵育5分钟。将管以8000rpm离心1分钟,弃去含有滤液的试管。将qiaamp旋转柱置于干净的2ml收集管中。向每个柱中加入500μl缓冲液aw2而不润湿边缘。盖上盖子,将离心管以14000rpm离心3分钟。从柱子中取出尼龙/棉拭子。将qiaamp旋转柱置于新的收集管中并以14000rpm离心3分钟。弃去含有滤液的离心管。将qiaamp旋转柱置于干净的,标记的1.5ml收集管中,并将样本在60℃下孵育20-25分钟,且盖子打开。向每个柱中加入50μl缓冲液ae(预热至65℃),并将样本在室温下孵育5分钟。将管以8000rpm离心以从柱中洗脱dna,并将样本在4℃储存。实施例2:膀胱样本dna提取a)从尿液样本中提取dna通过从对照和测试受试者收集尿液获得膀胱样本。将尿液样本平衡至室温(15-25℃)。旋转样本,将hemastix条带快速浸入尿液中以测试血液的存在。一分钟后读取hemastix测试结果并记录。将尿液样本转移至标记的50ml锥形管中,并以3500rpm离心2分钟。除去上清液,从锥形管的底部测量沉淀球团的长度。如果从锥形管底部测量沉淀球团长度小于2mm,则加入250μldna级水以悬浮该沉淀球团。如果测量的沉淀球团长度超过2mm,则加入1mldna级水以悬浮颗粒。涡旋样本,将250μl每种样本等分至1.5ml微量离心管中。如果样本超过250μl,则将多余样本保存在-18±2℃。记录用于悬浮沉淀球团的尿液和水的总体积,并将等分样本储存在-18±2℃直至用于提取。在室温下解冻样本,并向每个250μl等分样本中加入1120μl缓冲液avl/载体rna。将样本涡旋15秒并在室温(15-25℃)下孵育10分钟。将每个等分样本分装到2个微量离心管(每个685μl)中,并向每个管中加入560μl96-100%乙醇。将离心管涡旋15秒以除去盖子内部的液滴。将来自每个样本的第一个微量离心管的622μl溶液加到相应的qiaamp旋转离心柱,将管以>8000rpm离心1分钟,并弃去滤液。将来自每个样本的第一个微量离心管的剩余623μl加入相应的柱中,将管以>8000rpm离心1分钟,并弃去滤液。将来自每个样本的第二个微量离心管的622μl溶液加到相应的qiaamp旋转离心柱,将管以>8000rpm离心1分钟,并弃去滤液。将来自每个样本的第二个微量离心管的剩余623μl加入到相同的柱中,将管以>8000rpm离心1分钟,并弃去滤液。向旋转柱中加入500μl缓冲液aw1,将管以>8000rpm离心1分钟,并弃去滤液。向旋转柱中加入500μl缓冲液aw2,将管以全速(≥14000rpm)离心3分钟,并弃去滤液。将管再次以全速离心1分钟。将qiaamp旋转柱置于微量离心管中,并将30μl预热(70℃)dna级水加入柱中。将样本在室温(15-25℃)下孵育5分钟。将管以>8000rpm离心2分钟以从柱中洗脱dna,将洗脱液低温真空离心干燥20分钟直至液体蒸发,并将dna溶解于30μldna级水中。将dna样本在-18℃±2℃下短期储存(最多六个月)或在≤-65℃下永久保存。使用10%漂白剂清洗工作区域,然后用去离子水清洗。d)尿液dna定量quantifilertm人dna定量试剂盒用于定量人基因组dna。该试剂盒基于使用两种特异性5′核酸酶测定的taqman技术。一种是针对总人dna或y染色体(男性)的靶特异性测定,第二种是内部pcr对照(ipc),其检测由于过多dna或常见pcr抑制剂引起的抑制。ipc组分由ipc模板dna,自然界中未发现的合成序列,两个引物和一个vic标记的探针组成。靶特异性测定组分由用于扩增靶dna和taqmanmgb探针的两个引物组成。其中含有与5′末端偶联的6-羧基荧光素(fam)报告染料和带有与3′末端偶联的非荧光淬灭剂(nfq)的小沟结合剂(mgb)。mgb增加探针的解链温度而不增加其长度。quantifilertm人dna定量引物和探针靶向位于染色体位置5p15.33的人端粒酶逆转录酶基因(htert);扩增子长度为62个碱基。quantifiler-ytm定量引物靶向染色体位置yp11.3的y基因的性别决定区(sry);扩增子长度为64个碱基。在pcr过程中,taqman探针在上游和下游引物之间的特定区域退火。当探针完整时,淬灭剂足够接近报告分子以抑制报告荧光。当dna-聚合酶通过引物延伸时,报告分子从探针上被切下,导致荧光增加。荧光增加与产生的pcr产物的量成比例。对于总dna和y特异性dna来说,quantifilertm测定用于通过从标准曲线插入它们的量来定量未知样本。该测定可以准确地定量63pg至100ng的人dna。e)制备步骤用10%漂白剂对所有支架,台面和设备进行净化去污并彻底干燥。依次用10%漂白剂,水,100%甲醇清洗器械和移液管,并彻底干燥。确保所有设备在适当情况下符合质量控制的最低标准。从扩增后房间转移到扩增前房间的任何供应品或设备在取出前用10%漂白剂灭菌,并在使用前再次在扩增前房间中灭菌。为了尽量降低测试和匹配的对照样本之间交叉污染的可能性,不会将试剂从扩增后房间转移到扩增前房间。不将等分试剂放回原始储备容器。每天新鲜制备quantifilertm人dna标准品并在5℃±3℃下储存。quantifilertm试剂盒提供的dna储备溶液为200ng/μl。以下量用于创建标准曲线:50ng/μl,16.7ng/μl,5.56ng/μl,1.85ng/μl,0.620ng/μl,0.210ng/μl,0.068ng/μl和0.023ng/μl。向每个管中加入适量的无菌水和quantifilertm人dna标准品,并将管涡旋30秒以充分混合。用2μl无菌水代替模板dna制备阴性(无模板)对照。从冰箱中取出适当的引物混合物,解冻,涡旋混合并短暂离心。将quantifilertmpcr反应混合物从冰箱中取出,并上下吹打以混合均匀。pcr反应混合物制备如下:*n反应孔的总数以及1.1是过量10%。将23μl反应混合物等分到所有孔中。涡旋样本后,将2μl每个标准曲线样本加入96孔板的适当测试孔中。在添加完所有标准曲线样本后,将2μl阴性(无模板对照-水)加入到适当的孔中,并将2μl未知样本加入适当的测试孔中。用胶带覆盖该板,涡旋,并轻敲以除去气泡。或者,将样本板以3000-4000rpm离心2分钟,使内容物到达孔的底部并除去气泡。将平板置于abiprism7500和7500快速实时pcr系统上进行分析。c)实时pcr分析通过比较其信号强度与人dna标准对照产生的标准曲线的强度,并将该数除以2得到ng/μl结果,来测定每个样本中存在的dna的量。检查标准曲线结果以评估定量标准反应的结果质量。标准曲线是相对于标准品的起始量绘制的定量标准反应的ct图。该软件通过计算与量化标准数据点的最佳拟合来计算回归线。r2值是标准曲线回归线与定量标准反应的各个ct数据点之间的拟合紧密度的量度。数值为1.00表示回归线和数据点之间的完美拟合。确保r2值>0.98。如果r2值小于0.98,则检查以下内容:1)定量标准品系列稀释液的制备,2)定量标准品的反应加载量,和3)含有定量标准品的反应失败。斜率接近-3.3表示最佳(100%)pcr扩增效率。quantifiler人试剂盒的典型斜率范围是-2.9至-3.3,平均值为-3.1。如果标准曲线不符合参考,则可以去除不超过两个异常数据点,将这些孔指定为未使用。去除孔板文件中与异常数据点对应的孔,并重新分析该孔板以纳入该变化。如果去除这些数据点没有改善标准曲线,则定量被认为是无效的并且必须重复。阳性对照为实际浓度的±10%。实施例3:微卫星基因座的扩增a)用于msa的最佳dna量每个尿液样本提取的dna量决定了如何进行测定实验。最佳地,尝试使用两个多重pcr反应来进行完整测定。通过使用每个多重反应物中dna标称浓度的一半,在15μl水中低至0.5ng/μl的浓度可用于测试所有多重反应物多重反应物。在提取的总dna小于8ng的情况下,使用以下参考进行测定:如果从尿液样本中获得2.00-3.99ngdna,则在pcrl、2和3中每个使用2ngdna;如果从尿液样本中获得4.00-6.99ngdna,则在pcr1、2和3中每个使用3.5ngdna;如果从尿液样本中获得7.00-8.99ngdna,则在pcr1、2和3中每个使用4ngdna。无论是否有更多的颊dna,都将相同量的dna用于颊样本分析和尿液样本分析。在相同的实验中,在相同的热循环仪上,在相同的条件下,同时pcr扩增患者配对的颊和尿液dna。b)用于msa的pcr:引物序列用于msa测定的pcr引物对如表1所示。设计三种多重反应物(mpl,mp2,mp3)以扩增14种靶标。c)用于msa的pcr:pcr引物反应混合物对于每种引物测定反应混合物,将各引物稀释至2pmol的最终工作浓度。在pcr1中扩增的基因座是d4s243、fga、d9s747、d17s654和d17s695。在pcr2中扩增的基因座是d9s162、mbp、ifn-a、tho1和d16s310。在pcr3中扩增的基因座是d21s1245、d20s48、d9s171和d16s476。对于重复单重反应,通过组合87.5μldna级水和6.25μl上游和下游引物将引物稀释至5μm。对于每个引物混合物,制备100μl引物测定反应混合物,且按比例放大以制备更多等分试样。对于pcr1,将145μlpcr级水加入微量离心管中。添加5μl针对d9s747、d17s64和d17s695的上游和下游引物。在单独的微量离心管中,通过向5μlpcr级水中加入5μl引物从而将d4s243的上游和下游引物稀释2倍,并将5μl稀释的上游和下游引物加入到引物混合物中。将7.5μlfga的上游和下游引物加入到引物混合物中,并将引物反应混合物充分混合。制备一次性使用的30μl引物测定反应混合物的等分试样。对于pcr2,将150μlpcr级水加入微量离心管中。加入5μl用于针对d9s162、mbp,ifn-a的上游和下游引物。在单独的微量离心管中,通过向5μlpcr级水中加入5μl引物从而将d16s310和tho1的上游和下游引物稀释2倍,并将5μl稀释的上游和下游引物加入到引物混合物中,并将引物反应混合物充分混合。对于pcr3,将150μlpcr级水加入微量离心管中。添加5μl针对d21s1245和d20s48的上游和下游引物。在单独的微量离心管中,通过向5μlpcr级水中加入5μl引物从而将d9s171和d16s476的上游和下游引物稀释2倍,并将5μl稀释的上游和下游引物加入到引物混合物中,并将引物反应混合物充分混合。d)用于msa的pcr:pcr将试剂体积乘以加上阳性和阴性对照的样本数目以计算反应混合物所需的体积。对于每十个样本,添加10%以用于补偿移液过程中的体积损失。将批号记录在msa扩增工作表中。按顺序组合以下试剂,以产生pcr1、2和3的反应混合物:12.5μl2xfaststarttaqmanprobemaster,1μl引物反应混合物(1、2或3)和7.5μl水,总计21μl。将21μl反应混合物等份加入到每个孔中用于pcr1和pcr2。对于对照反应,将4μl(多重反应物1、2、3)对照dna加入孔中作为阳性对照,并将4μl(多重反应物1、2、3)水加入孔中作为阴性对照。对于每个患者样本反应,将4ng(4μl)dna加入到pcrl、pcr2和pcr3中。对于通过单重pcr重复反应来分析的患者样本,将1ng(1μl)dna加入到pcr1和pcr2中。将pcr板(或96孔pcr支架中的pcr管)以3000rpm短暂离心约1分钟,将所有内容物带到板底部。pcr条件如下:95℃11分钟一个循环,94℃1分钟32个循环,55℃1分钟和72℃1分钟,60℃45分钟一个循环,最后保持在4℃。然后将样本储存在-4℃下短期储存或在-18℃下长期储存。用10%漂白剂清洗pcr仪器区域,然后用水擦拭并打开uv灯照射5分钟。实施例4:通过abi3130遗传分析仪鉴定人类dnaa)用于遗传分析仪pcr反应的样品的制备制备含有甲酰胺(8.7μl)和liz(0.3μl)大小标准量(每孔总共9.0μl)的样品上样缓冲液。板柱中所有未使用的孔用10μl甲酰胺或liz反应混合物填充,并且相邻偶数柱中的所有孔用10μl甲酰胺填充。将1.3μl的各pcr反应物加入到样品板的适当孔中,并将样品板进行短暂离心以将内容物带到各孔底部并除去气泡。将样品置于加热块上,在95℃条件下加热3分钟,然后置于冷却板的冰上(-18±2℃),或置于4℃的热循环仪中持续2分钟。通过在样品板中插入隔片,将样品板组装以备使用,将样品置入板底座并夹紧在板的保持器上。位置1的缓冲液槽装有新鲜的1x遗传分析缓冲液(通过以1∶10的比例稀释10x的3130缓冲液edta获得),位置2-4的槽装满水。abi3130遗传分析仪以每个分析文件夹以两列为一组降序16个孔分析片段数据。患者配对的颊和尿液样品在同一次电泳中运行且在16个孔分析的同一文件夹中。b)设置遗传分析仪软件根据本发明开发的“py软件”基于abi3130遗传分析仪。具体操作为:打开genemapperid3.2版或更高版本,输入用户id(gmid)和密码,选择“tools”按钮下的“panelmanager”,选择“file”下的“newkit”,选择“microsatellite”并命名该panel为“pytestpanel”,单击ok,将箭头放在名为“pytestkit”的新kit上,选择“file”下的“newpanel”,选择“bins”下的“newbins”并为其进行命名,选择“file”下的“newpanel”,重命名该panel为“multiplex1”,选择multiplex1,然后输入图表中每个标记物的信息。重复相同的步骤设置multiplex2和multiplex3。为设定分析方法,在“tools”下选择“genemappermanager”,选择“analysismethodeditor”,然后选择“new”,通过“general”选项将analysismethodeditor命名为“pysop”,并在“allele”选项中的binset下选定“pytest”。选择“rangerfiler”按钮,设置红色通道用于删除从186.5到188的标记。设置峰值检波器(peakdetector),峰值质量(peakquality)和质量按钮(qualitytabs)。通过选择“tools”下的“genemappermanager”,再选择“sizestandard”选项,选择并打开“gs500lizstandard”来设置尺寸标准。应输入并保存标准值100,139,150,160,200,300,340,350,400,450,490,500。设置3130分析仪的设备方案:打开3130数据收集软件,选择“modulemanager”并选择“new”,选择“fragmentanalysis36_pop4:”作为模板,为该模块创建新名称,例如fragementanalysis36_pop4_1。通过如下操作设置运行模式(runsettings):烘箱温度60℃,预运行电压15,预运行时间180秒,注射电压3.1千伏,注射时间8秒(或样品重新注入时间5秒),电压步骤数量10nk,电压步骤间隔60秒,数据延迟时间1秒,运行电压15伏,运行时间1500秒。选择“protocolmanager”,选择“new”,创建方案名称,例如“newtestprotocol”,将类型选定为“regular”,将3130分析仪的运行模块设置为上面创建的模块,并将dyeset设置为g5。为制作样品表,选择“appliedbiosystems-datacollection-rundatacollector2.0”,打开ga3130文件夹,选择“platemanager”,选择“option”,选择“new”,输入并在msapcr扩增工作表上记录所述板的名称。依据应用程序和所有者名选定“genemapper-(nameofcomputer)”,并填写操作员名称。在genemapper板编辑器屏幕上,输入样品名称,优先级设置为100,选择样品类型,尺寸标准被设置为测试标准模式,根据运行的多重复合物,选择测试面板/多重复合物1(pcr1)或多重复合物2(pcr2),分析方法被设置为“choosenewtest”,跳过标题为snp的部分,设置用户定义1,2和3,结果组1被设置为“str”,设备方案1设置被为“testprotocol”。为执行运行,打开标题为“3130”的文件夹,选择“runscheduler”,选择“findall”,选定目标运行,突出显示所需的板,并开始运行。运行2个泳道大约需要45分钟,当运行完成时,取出板,用costarthermowell密封剂密封,并在-18±2℃下储存至1周。4-c:使用py软件分析msa数据将数据导入genemapperid3.2版或更高版本中,并由其程序进行分析。必要时对样品进行手动编辑。对于手动编辑sq列具有红色或黄色标记的样本时,应选择大小匹配的编辑器(依据大小标准峰确定),并通过点击“overridesq”和“apply”手动更改不正确的峰的大小,然后重新分析样品。c)确定msa峰值大小每个分析包括配对的患者样本的口腔dna和尿dna的数据。这些是一并分析的(即两样品同时进行pcr1,两样品同时进行pcr2,和两样品同时进行pcr3)。要调整大小的两个样品被突出显示,并选择“displayplots”。通过比较每个多重反应物的口腔样品和尿液样品来查阅这些作图。对于每个样品和每个多重反应物,每个通道(蓝色,红色,黄色,绿色)都被查阅。对于分配峰特定等位基因的考虑至此完成。图1-3示出了ifn-α,d9s171和d16s476的峰图的示例。完成大小调整后,选择“genotypetab”,选择“msaanalysis”模式,并将基因型信息用于验收标准的统计解读。d)关于3130遗传分析仪片段大小分析的解读每个样品的分析由一名分析员进行,数据输入由第二名分析员验证。获得genemapper的峰尺寸和高度数据表,通过使用超过40个经手术切除的膀胱癌样本及其对照组建立验收标准,具体如表2所示。表2微卫星标记物分析基因座重复类型:尺寸范围(单位:bp)通道颜色k562~等位基因尺寸d4s243(atag)n165-192bp蓝色169bpfga(tttc)n299-361bp绿色328bpd9s747(gata)n179-201bp绿色185bpd17s654(ca)n194-218bp黄色216bpd9s162(ca)n117-148bp黄色143bpd17s695(aaag)n170-220bp红色185,200bpmbp&a(atgg)n200-242/119-151bp蓝色207,215,119bpd21s1245(aaag)n209-293bp绿色236,255bpd16s310(atag)n127-170bp绿色155,160bpd20s48(gt)n251-269bp黄色261bptho1(tcat)n174-209bp黄色198bpd9s171(ca)n109-129bp黄色126bpd16s476(aaag)n176-230bp红色187209bpifn-a(gt)n132-152bp红色无产物对于被称为“正常”的样品,其扩增产物必须符合表2中所示的尺寸大小(±1bp)和颜色,并且对于每个杂合等位基因具有最少100相对荧光单位(rfu),对于每个纯合等位基因具有最少200rfu。对于任何基因座,阴性对照不产生pcr产物(rfu<100)。对于被称为“癌症”的样品,其扩增产物必须符合表2中所示的尺寸大小(±1bp)和颜色。对于被称为“癌症”的口腔样品,其扩增产物对于每个杂合等位基因必须具有最少100相对荧光单位(rfu)、对于每个纯合等位基因必须具有最少200rfu。对于被称为“癌症”的尿液样品,其扩增产物必须具有至少一个等位基因的至少100相对荧光单位(rfu),并且对于被认为是等位基因的两个峰,其中一个等位基因的rfu必须在另一等位基因rfu值的40%以内。等位基因的最大rfu值为5000rfu。基于公布的数据和在此进行的分析,表3示出了用于确定loh的每个标记物边界值。表3标记物边界值(超出该范围=loh)标记物最低值最高值d4s2430.681.33fga0.601.43d9s7470.631.42d17s6540.711.36d17s6950.491.61mbp0.631.45mbpa0.711.37d16s3100.61.34d9s1620.511.53thol0.461.53ifn-a0.681.43d21s12450.581.42d20s480.621.46d9s1710.721.36d16s4760.541.65e)基于msa颊粘膜拭子的数据分析样品数据有序成对以用于分析。样品的降序如下:口腔1(含多重反应物1中每个基因座的结果),尿液1(含多重反应物1中每个基因座的结果),口腔2(含多重反应物2中每个基因座的结果),尿液2(含多重反应物2中每个基因座的结果),口腔3(含多重反应物3中每个基因座的结果),尿液3(含多重反应物3中每个基因座的结果)。hl60数据按如下排序:多重反应物1,多重反应物2和多重反应物3。基因座以与颊的顺序相同的顺序列出。电子表格的列标题如下:样本序号,基因座,等位基因1大小,等位基因2大小,(两个空列),等位基因1峰高,等位基因2峰高。将数据导入excel进行分析。将数据与上述验收标准进行比较,以确定数据是否通过验收标准。如果样本未通过验收标准,则不会在评估中使用。为了确定样本的状态,将数据输入excel电子表格(msasamplespreadsheet.xls)。该电子表格使用以下算式来确定口腔/尿液峰高度的比率:比率=(尿液1等位基因1峰高/尿液1等位基因2峰高)/口腔1等位基因1峰高/口腔1等位基因2峰高。通过使用基于每个基因座验收标准的算式,可以自动确定比率和msi或loh标记物的阴性/阳性。显示为loh(通过等位基因的丢失)的基因座被认为是loh阳性,尽管显示为完全丢失的基因座比率无法计算得出。该比率仅针对杂合性基因座和具有完整数据组的基因座进行计算。将该比率与边界数据进行比较,落在边界范围之外的比率被认为是loh阳性,落在该范围内的比率被认定为msi阴性或loh阴性。如果在尿液中检测到额外的或移位的等位基因,则该额外的或移位的等位基因在箱内移位±3bp且该额外的或移位的等位基因具有100rfu的最小峰高,则确定该基因座为msi阳性。f)3130遗传分析仪所得结果的评估:重复标准测试当有足够的剩余dna进行测定时,可建立重复标准用于重复测试。如果没有足够的dna,则认为原始运行的结果对于满足测定验收标准的所有基因座都是有效的并且被报告。如果整个hl60多重反应物阳性对照不能满足验收标准,则不对含有未通过验收标准的多重反应物的样本数据进行评估,且在原dna浓度下对合适的hl60和样本多重反应物进行重复测试。如果hl60对照多重反应物中有一个或多个基因座未能满足验收标准,则不评估这些未通过验收标准的基因座的样本数据。在原dna浓度下对合适的hl60和样本多重反应物进行重复测试,且在msasamplespreadsheet中仅报告第一次运行期间未通过验收标准的基因座的结果。第一次运行期间通过验收标准的基因座的数据在第二次运行的样本表中被移除。如果整个样本多重反应物未通过验收标准,在原dna浓度下对该多重反应物进行重复测试。如果样本具有单个的阳性结果的基因座,则使用1ng的dna和5μm的各引物以单重形式对该基因座进行重复测试,并报告两种测试的结果。按上述方式,以单重形式对具有所指的微卫星不稳定性的额外的尿液峰的基因座进行重复测试,以确认该额外的峰。如果基因座内的任何等位基因具有大于5000(超出正常范围)的峰高,则通过重新注射多重反应物(注射时间为5秒)对该基因座进行重新评估。决策流程图用于确定在重新注射的评估中包括哪个基因座。如果纯合等位基因不干扰对其他基因座的评估的能力,则这些峰高大于5000的纯合等位基因不会被重新注射。如果使用重新注射值,则报告重新注射的样本的口腔和尿液值来代替第一次运行中获得的超出正常范围或模糊的峰的口腔和尿液值。如果(峰值)仍然超出正常范围或模糊,则结果为不可评估。g)样本结果的最终报告在所有重新注射或重复结果之后,在含多重反应物数据的原始样本电子表格上报告样本的每个基因座的最终确定结果。实施例5:临床试验——通过尿液沉渣的msa检测膀胱癌a)样本“pysop”和py测试用于预测人生物样本中存在或缺少膀胱癌。分析来自“正常”患者的5对配对的颊粘膜拭子和尿液样本以及来自膀胱癌患者的5对配对的颊粘膜拭子和尿液样本,以及阳性和阴性dna样本对照和无模板阴性对照。每个样本由指定的clia测试实验室(dctl)样本编录组分配的唯一标样id进行鉴定。所有样本均按照cap和clia规定进行处理,确保在整个测试过程中跟踪样本。b)msa的pcr进行msa测定。该测定由对匹配的颊粘膜拭子和尿沉渣基因组dna进行dna提取、标准化、str标记物的pcr扩增组成。使用位于14个微卫星基因座的靶str侧翼的引物组进行pcr。每个引物对的5′末端进行荧光标记,以允许通过毛细管电泳检测pcr片段。前述表1中的引物对可供使用。在基于毛细管的凝胶电泳系统上分辨pcr扩增子,该系统可检测、测量和确定每个片段的相对荧光单位(rfu)。将在颊粘膜拭子dna中检测到的杂合等位基因的rfu与在配对的尿液样本中检测到的rfu进行比较,并计算来自尿液等位基因的rfu与血液等位基因的比率。显示出在正常样本中所见比率之外的值的标记物则被认为存在杂合性丢失(loh),该杂合性丢失可作为膀胱癌的标志。示例性电泳图,请参见图4。c)msa测定对照存在用于计算每种pcr产物片段大小的尺寸标准的情况下,对每个样本和对照组进行电泳。阳性对照基因组dna来自美国模式培养物研究所(atcc)和appliedbiosystems。对照的测试在低于由样本临床验证中确定的检测下限的浓度条件下进行。d)仪器和软件用于测定的仪器是appliedbiosystems的3130xl遗传分析仪,能够在1小时内产生基因分型数据的16毛细管仪器。3130xl基因分析仪可处理各种基因检测分析,包括dna测序,snp基因分型和片段分析。3130xl基因分析仪采用genemapper软件2.0版运行,该软件符合美国联邦法规第21篇第11部分的规定。根据cap规定,在光谱和空间校准方面,该仪器每半年校准一次。e)样本采集和制备收集两种样本:颊粘膜拭子和尿液沉渣。颊粘膜拭子由指定的clia测试实验室(dctl)使用收集套件收集,并遵循明确的收集说明。简言之:(1)颊粘膜拭子在4℃稳定一周;(2)一旦在dctl被收到,每个案例都被编录并分配一个唯一的标识符,该标识符将在整个提取过程中跟随样本;(3)在cap认可的实验室中从样本中提取基因组dna。使用appliedbiosystems的quantifilerreal-timepcr试剂盒对样本中人类dna进行定量,并将基因组dna标准化为1ng/μl。通过dctl使用收集试剂盒并按照明确的收集说明收集尿液沉渣。简言之:(1)将尿液收集到无菌收集杯中,并加入防腐剂以稳定尿液中的dna,将尿液和防腐剂混合,并将尿液核酸和蛋白质在室温下保存以便运输;(2)使用norgenbiotech的urinednaisolationmaxi试剂盒,从尿液沉渣中纯化出基因组dna,该试剂盒从25ml至80ml的尿液中分离出高分子量dna(大小超过lkb;大部分与细胞结合)和小分子量dna(150-250bp;来自循环系统)。使用appliedbiosystems的quantifilerreal-timepcr试剂盒对样本中人类dna进行定量,并将基因组dna标准化为1ng/μl。从5名被诊断患有膀胱癌的患者和匹配的5名正常/健康患者中获得匹配的尿液和颊粘膜拭子样本组。样本获自bioreclaimationllc。从正常患者收集两个颊粘膜拭子,从癌症患者收集一个颊粘膜拭子。通过生物再生(bioreclaimation)对样本去识别化,去除任何hippa敏感信息,并通过生物再生(bioreclaimation)为每个样本指定样本id。图5中提供了每个样本的生物特征数据,具体样本编号由表4示出。表4用于分析的样本编号f)从样品中提取dna将尿液样品进行离心操作以沉淀细胞。轻轻倒出尿液,将沉淀球团重新悬浮在水中。将250μl尿液沉渣浆液移液到微量管中,保留剩余的尿液沉渣并储存在-20℃±2℃环境中。向每个样品中加入缓冲液avl/载体rna。将样品涡旋并在室温下温育10分钟。遵循qiaampviralrna迷你试剂盒说明书进行基因组dna提取。g)dna定量和标准化使用实时pcr(taqman定量pcr)、通过od260确定的1ngdna的直接测试来确定提取样品中人类dna的浓度(参见cbidnaseq00035)。人类β肌动蛋白基因是taqman测定的靶标。具有已知浓度的人标准dna的标准曲线被用于与来自样品提取物的结果进行比较,以确定浓度。taqman测定甚至可以确定浓度的微小差异,并且对10个拷贝的dna分子很敏感。一旦确定样品dna浓度,样品将被标准化至1ng/μl。将样品中dna浓度示于表5中。表5:患者样品的dna浓度h)msa的pcr和分析使用amplitaqgold作为酶,如实施例3中所述,进行msa的pcr。使用实施例3中描述的和表2中示出的验收标准。如实施例4中所述进行分析,并使用表3示出的边界值检测loh和msi。使用实施例4的f中给出的标准对loh或msi进行分类。i)结果表6展示了bioreclamation提供的样品对id及其相应的尿液和颊粘膜拭子样品id。表6:来自bioreclamationllc的样品对id和对应的样品id图6a-6e分别示出了表6中定义的样本对id:n1-n5的原始msa数据。图6f-6j分别示出了表6中定义的样本对idc1-c5的原始msa数据。表7显示了已运行的msa的结果,其中“n”代表正常(无loh),“loh”代表配对的颊和尿液样品之间存在杂合性丢失,“msi”代表配对的颊和尿液样品观察到了微卫星不稳定性。如果少于两个基因座表现出loh或msi,则“最终结果”被认为是“n”,或如果两个或多个基因座表现出loh或msi,则“最终结果”被认为是“c”。表7:msa结果汇总总之,通过临床诊断和膀胱镜确认的样本对通过本申请提出的msa方法确定的疾病是呈阳性的。对于测试的所有14种标记物,每个正常对照组对msi和loh均呈阴性。对于膀胱癌样本,八种特定标记物中的至少两种对loh呈阳性。所述八种标志物是fga,d98747,mbp,d98162,tho1,ifn-a,d21s1245和d20s48。因此,此处提供的基于颊的msa分析仅需要使用八种标记物来准确检测膀胱癌。本领域技术人员将理解,可以对本发明的优选实施例进行许多改变和修改,并且可以在不脱离本发明主旨的情况下进行这些改变和修改。因此,本申请的权利要求旨在覆盖落入本发明的真实主旨和范围内的所有这些等同变化。本文件中引用或描述的每个专利、专利申请和出版物的公开内容通过引用整体并入本文。序列表<110>bcd创新公司<120>在配对的颊粘膜拭子和尿液样本中使用微卫星分析进行膀胱癌检测<130>099916.000016<140><141><150>62/264504<151>2015-12-08<160>28<210>1<211>22<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>1tcagtctctctttctccttgca22<210>2<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>2taggagcctgtggtcctgtt20<210>3<211>23<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>3gacatcttaactggcattcatgg23<210>4<211>23<212>dna<213>日本柳杉<400>4cttctcagatcctctgacactcg23<210>5<211>24<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>5gccattattgactctggaaaagac24<210>6<211>25<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>6caggctctcaaaatatgaacaaaat25<210>7<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>7acctaggccatgttcacagc20<210>8<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>8gagcagaatgagaggccaag20<210>9<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>9ctgggcaacaagagcaaaat20<210>10<211>26<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>10tttgttgttgttcattgacttcagtc26<210>11<211>22<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>11gcaaccatttatgtggttaggg22<210>12<211>21<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>12tcccacaacaaatctcctcac21<210>13<211>23<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>13ggacctcgtgaattacaatcact23<210>14<211>24<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>14atccatttacctacctgttcatcc24<210>15<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>15gggcaacaaggagagactct20<210>16<211>22<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>16aaaaaaggacctgcctttatcc22<210>17<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>17aggctctagcagcagctcat20<210>18<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>18tgtacacagggcttccgagt20<210>19<211>21<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>19tgcgcgttaagttaattggtt21<210>20<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>20gtaaggtggaaacccccact20<210>21<211>22<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>21ccagaaaatgacacatgaagga22<210>22<211>21<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>22ttgttgaggatttttgcatca21<210>23<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>23atggtctccagtcccatctg20<210>24<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>24ttgacctggatgagcatgtg20<210>25<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>25tctgtctgctgcctcctaca20<210>26<211>22<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>26gatcctatttttcttggggcta22<210>27<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>27ggcaacaagagcaaaactcc20<210>28<211>20<212>dna<213>人工序列<220><223>人工序列的描述:合成引物<400>28ggtgctctctgccctatctg20当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1