一种车前抗氧化作用成分的提取方法与流程
本发明属于农业领域,具体涉及一种车前草科植物的提取方法。
背景技术:
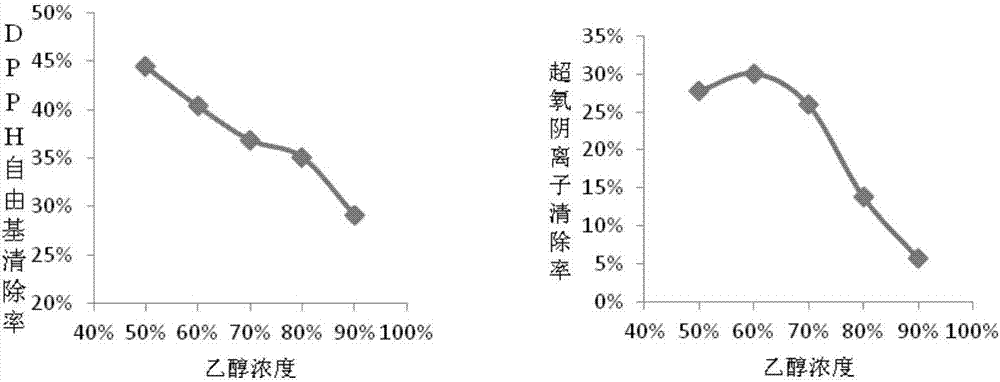
:畜禽在养殖过程中由于遗传选择、饲料原料、环境等原因,极易受到氧化应激,影响畜禽生长。合成抗氧化剂因其成本低、活性好,在饲料生产、食品等方面应用广泛,但同时合成抗氧化剂也存在着健康问题、致癌风险、产品残留及合成过程中的副反应污染问题。天然抗氧化剂因其具有使用安全和潜在的功效等活性、特点,有利于畜禽健康养殖,因其天然来源更为消费者所接受。车前草,又名平车前、车轮草等,为二年生或多年生草本,在我国大部分地区有分布,具有适应性强、耐寒、耐旱等特征。车前属植物在世界范围内大约有275种,作为传统药物在世界范围内有着不同的用途,其中20种广泛分布在我国境内,资源丰富。大车前、车前、平车前全株及其籽自古以来在我国就一直作为食品和传统医药使用。车前为车前或平车前的全草干燥制成,含有黄酮类、多糖类、环烯醚萜类、苯乙醇苷类、三萜类等多类化学活性成分。车前作为传统医药长期以来用于促进伤口愈合,用于皮肤及其他感染、消化和呼吸道紊乱的治疗,可改善循环、繁殖,缓解疼痛及发热。已有肉鸡饲养的试验表明,车前对肉鸡的氧化还原状态表现出良好的改善效果,但在饲料中直接添加车前,则因其有效成分低导致添加剂量过高,限制了车前的推广应用。技术实现要素:针对本领域的不足之处,本发明的目的是提供一种车前抗氧化作用成分的提取方法。本发明的第二个目的是提出所述提取方法得到的产物。实现本发明上述目的的技术方案为:一种车前抗氧化作用成分的提取方法,包括步骤:1)车前粉碎,按照料液比1:5~30(克/毫升)加入乙醇溶液进行提取,提取的方式为超声提取;2)对步骤1)得到的混合物进行固液分离,取清液,去除溶剂。其中,车前为车前或平车前草干燥的全草。其中,步骤1)中,车前粉碎的粒度为20~200目;所述乙醇溶液的质量浓度为50~85%。车前粉碎的粒度优选为120~160目。其中,步骤1)中,一次超声提取的时间为20~40min,共进行2~5次,优选超声3次。其中,步骤2)去除溶剂的操作在10~40℃温度下进行,去除溶剂的方式为旋转蒸发、自然干燥、或真空干燥中的一种。进一步地,所述的提取物方法,还包括对步骤2)所得提取车前提取物的分离步骤:采用硅藻土固相萃取、硅胶层析、凝胶和HPLC色谱分离方法中的一种或多种从车前提取物中分离出黄酮类化合物和酚酸类化合物;所述硅藻土固相萃取的溶剂按顺序为石油醚、二氯甲烷、乙酸乙酯、乙酸乙酯和甲醇混合溶剂、甲醇顺序,进行连续固液萃取。其中,所述分离步骤中,乙酸乙酯和甲醇混合溶剂按体积比分别为10:1、5:1、2:1,按乙酸乙酯体积递减的顺序连续固液萃取,取乙酸乙酯部位和乙酸乙酯甲醇混合溶剂体积比10:1部位的浸膏;乙酸乙酯部位的浸膏经硅胶柱层析,以二氯甲烷:甲醇体积比100~5:1梯度洗脱,得黄酮类化合物;乙酸乙酯和甲醇混合溶剂体积比10:1部位的浸膏经硅胶柱层析,以二氯甲烷:甲醇体积比20~2:1梯度洗脱,得酚酸类化合物。更进一步地,所述乙酸乙酯部位的浸膏经硅胶柱层析,其中二氯甲烷:甲醇体积比25~35:1洗脱的流份经凝胶柱层析后,通过高效液相色谱(HPLC),用甲醇水体系等度洗脱,得到单体甘草素(P1);其中二氯甲烷:甲醇体积比18~25:1洗脱的流份,经凝胶柱层析得到芹菜素单体(P2)。其中HPLC等度洗脱的流动相可以为55%甲醇:水(即甲醇的体积分数为55%)。所述乙酸乙酯和甲醇混合溶剂体积比10:1部分的浸膏经硅胶柱层析,取二氯甲烷:甲醇体积比10:1洗脱的流份重结晶得七叶内酯(P6),取二氯甲烷:甲醇体积比2:1洗脱的流份;经ODS柱洗脱,再经HPLC分离得到4,5-二咖啡奎宁酸甲酯(P5)。优选地,分离4,5-二咖啡奎宁酸甲酯过程中,所述ODS柱按30%-100%甲醇:水梯度洗脱;所述HPLC的流动相为40%-90%甲醇:水,流速为18~20ml/min。本发明所述提取方法获得的产物。本发明的有益效果在于:(1)本发明提出的车前抗氧化物提取工艺,较常规浸泡法相比节省溶剂和提取时间,提取效率更高。(2)本提取工艺获得的车前提取物具有良好的抗氧化活性。附图说明图1为料液比和去除自由基能力的关系图。图2为乙醇浓度和去除自由基能力的关系图。图3为提取次数和去除自由基能力的关系图。图4为提取时间和去除自由基能力的关系图。图5为粉碎粒度和去除自由基能力的关系图。图6为P1甘草素的H-NMR图谱。图7为P1甘草素的C-NMR图谱。图8为P2芹菜素的H-NMR图谱。图9为P54,5-二咖啡奎宁酸甲酯的H-NMR图谱。图10为P6七叶内酯的H-NMR图谱。图11为4,5-二咖啡奎宁酸甲酯的HPLC分离流出曲线。图12为甘草素的HPLC分离流出曲线。图13为分离P1、P2化合物的流程图。图14为分离P5、P6化合物的流程图。具体实施方式现以以下实施例来说明本发明,但不用来限制本发明的范围。实施例中使用的手段,如无特别说明,均使用本领域常规的手段。实施例中车前为车前或平车前的全草干燥制成。实施例1:车前抗氧化物质提取影响因素确定选取料液比、乙醇浓度、粉碎粒度、提取次数、提取时间、旋转蒸发温度5个因素进行单因素试验,以确定最佳提取条件,试验方法如下:1.1料液比对提取物清除DPPH自由基和超氧阴离子自由基的影响:将车前草粉碎过20目筛,精确称取5.0g样品,按照不同的料液比(1:5、1:10、1:15、1:20、1:25、1:30,单位为g/mL)分别加入80%(质量浓度)的乙醇水溶液,超声40kHz提取30min。过滤样品溶液,旋转蒸发除去乙醇(<40℃),减压浓缩后定容并测定其清除DPPH自由基(图1左图)和邻苯三酚自由基(图1右图)的能力,重复三个平行。结果见图1。以5.0g车前原料计,提取物对32mg/ml(处理前的初始含量,下同)DPPH自由基的去除率可达29%,对60mmol/l…邻苯三酚自由基的清除率可达14%。1.2乙醇浓度对提取物清除DPPH自由基和超氧阴离子自由基的影响:将车前草粉碎过20目,精确称取2.0g样品,分别加入50ml70%、80%、90%、100%的乙醇,超声提取30min。4000转离心15min,取出上清液,旋转蒸发除去乙醇(<40℃),减压浓度后定容并测定其清除DPPH自由基(图2左图)和邻苯三酚自由基(图2右图)的能力,重复三个平行。结果见图2。以2.0g车前原料计,提取物对32mg/mlDPPH自由基的去除率可达45%,对60mmol/l邻苯三酚自由基的清除率可达30%。1.3提取次数对提取物清除DPPH自由基和超氧阴离子自由基的影响:将车前草粉碎过20目,精确称取2.0g样品,加入50ml80%的乙醇,分别超声提取1、2、3、4次。4000转离心15min,取出上清液,旋转蒸发除去乙醇(<40℃),减压浓度后定容并测定其清除DPPH自由基和邻苯三酚自由基的能力,重复三个平行。结果见图3。以2.0g车前原料计,提取物对32mg/mlDPPH自由基的去除率可达33%,对60mmol/l邻苯三酚自由基的清除率可达22%。1.4提取时间对提取物清除DPPH自由基和超氧阴离子自由基的影响:将车前草粉碎过20目,精确称取2.0g样品,加入50ml80%的乙醇,分别超声提取30、60、90、120min。4000转离心15min,取出上清液,旋转蒸发除去乙醇(<40℃),减压浓度后定容并测定其清除DPPH自由基和邻苯三酚自由基的能力,重复三个平行。结果见图4。以2.0g车前原料计,提取物对32mg/ml的DPPH自由基的去除率可达31%(图4左图),对60mmol/l邻苯三酚自由基的清除率可达4.5%(图4右图)。1.5粉碎粒度对提取物清除DPPH自由基和超氧阴离子自由基的影响:将车前草粉碎并过不同型号的筛子,分别精确称取20-80目、80-120目、120-160目、160-200目及过200目的车前草样品各2.0g,加入50ml80%的乙醇,超声提取30min。4000转离心15min,取出上清液,旋转蒸发除去乙醇(<40℃),减压浓度后定容并测定其清除DPPH自由基和邻苯三酚自由基的能力,重复三个平行。结果见图5。以2.0g车前原料计,对32mg/ml的DPPH自由基的去除率可达48%(图5左图),对60mmol/l邻苯三酚自由基的清除率可达8%(图5右图)。实施例2.车前抗氧化物质提取工艺正交优化在单因素试验结果基础上,选择乙醇浓度、粒液比、提取次数和粉碎粒度作为4个变量进行正交试验。正交设计表见下表1。为了对比超声提取和常规浸泡的差别,提取方法分别采用了超声提取和浸泡的方法。浸泡一次为12h,超声提取一次为30分钟。表1正交设计表乙醇浓度(%)料液比次数(次)粉碎粒度(目)5010280-12050203120-16050304160-2006010280-12060203120-16060304160-2007010280-12070203120-16070304160-200车前抗氧化提取物提取工艺正交试验结果如下表2所示:表2超声提取DPPH方法检测正交表及其结果其中,Ki代表各个处理的总和,ki代表各个处理的平均值,R代表各个处理的极差。下表同。表3超声提取邻苯三酚方法检测正交表及结果试验号A乙醇浓度(%)B料液比C次数D粉碎粒度(目)清除率(%)11(50)1(10)1(2)1(80-120)3.01212(20)2(3)2(120-160)8.82313(30)3(4)3(160-200)6.8842(60)1237.53522314.73623128.8273(70)1328.17832137.74933215.81K111.8318.7119.5713.55K221.0821.2922.1525.81K321.7221.5119.7822.15k13.946.246.524.52k27.037.107.388.60k37.247.176.597.38R3.300.930.864.09P值0.2430.2220.2920.331最优A2B2C1D1超声提取条件下,对DPPH自由基清除率的正交直观分析可以看出,四个因素对车前草粗提取物抗氧化能力的影响顺序为:乙醇浓度>粉碎粒度>次数>料液比,并得出该正交试验的最佳组合,即A1B2C2D2,即乙醇浓度为50%,料液比1:20,提取次数3次,粉碎粒度120-160目。对比例作为对比,用同样的车前样品作正交试验,得出浸泡提取的最佳组合为A2B3C3D3,即乙醇浓度为60%,料液比1:30,提取4次(浸泡4次,每次12小时),粉碎粒度为160-200目。正交试验设计和结果见表4和表5。在清除率差别不大的情况下(抑制率结果差别不显著),与常规浸泡相比,车前抗氧化提取工艺超声提取法较常规浸泡法能节省溶剂和时间。表4浸泡提取DPPH方法正交表及其结果试验号A乙醇浓度(%)B料液比C次数D粉碎粒度(目)清除率(%)1.001(50)1(10)1(2)1(80-120)39.782.0012(20)2(3)2(120-160)42.383.0013(30)3(4)3(160-200)48.0142(60)12344.485223147.226231243.3273(70)13237.768321335.389332139.06K1130.18122.02118.48126.06K2135.02124.98125.92123.47K3112.20130.40133.00127.87k143.3940.6739.4942.02k245.0141.6641.9741.16k337.4043.4744.3342.62R7.612.794.841.47P值0.000.020.000.26最优A2B3C3D3表5浸泡邻苯三酚方法检测正交表及其结果实施例3车前抗氧化活性物质成分分离按照优化的提取工艺提取车前抗氧化物质提取物,采用硅藻土固相萃取、硅胶、凝胶、HPLC等色谱技术从中分离得到并鉴定了4种化合物,其中黄酮类2个(P1、P2)、酚酸类2个(P5、P6),鉴定结果为如下化合物结构式,波谱分析图谱及HPLC流出曲线见附图6-12。单体物质的分离过程如下:(1)提取采用本申请中提取工艺:车前草中抗氧化物质的最佳提取条件是采用超声提取法,乙醇浓度为A1,料液比B2,提取次数C2次,粉碎粒度D2目。(2)萃取取粗提物(700g)用甲醇反复浸泡浸提至无浸出物,旋蒸浓缩得到浸膏(447.8g)。将甲醇浸提所得浸膏用甲醇溶解,按浸膏与硅藻土质量比1:1.3与硅藻土拌样后,按照石油醚、二氯甲烷、乙酸乙酯、乙酸乙酯:甲醇(10:1)、乙酸乙酯:甲醇(5:1)、乙酸乙酯:甲醇(2:1)、甲醇顺序按5倍柱体积进行连续固液萃取,分别得到石油醚部分浸膏15.61g、二氯甲烷部分浸膏12.53g、乙酸乙酯部位浸膏3.44g、乙酸乙酯:甲醇(10:1)部分浸膏148.34g、乙酸乙酯:甲醇(5:1)部分浸膏77.00g、乙酸乙酯:甲醇(2:1)部分浸膏30.25g、甲醇部分浸膏105.12g。(3)分离(3.1)乙酸乙酯部位共3.136g溶解后硅胶(5g,60-100目)拌样,经硅胶(56g,100-200目)柱层析,以二氯甲烷:甲醇(100:1、50:1、30:1、20:1、10:1、5:1;V/V)梯度洗脱,每浓度洗脱3个柱体积,共收集118流份,经薄层色谱检查合并为7份,分别为第1份(Fr.1-6),第2份(Fr.7-22),第3份(Fr..23-40),第4份(Fr.41-58),第5份(Fr.59-76),第6份(Fr.77-85),第7份(Fr.86-118)。第3份(F.3,0.383g)经硅胶柱层析,二氯甲烷:甲醇系统(100:1、50:1、30:1、10:1、8:1、5:1、2:1,每浓度5个柱体积)梯度洗脱分离得到68流份(图1-2)。薄层色谱点板分析合并34-43流份(F.3-6,0.042g),经硅胶柱层析(二氯甲烷:甲醇系统;100:1、50:1、30:1、10:1、5:1、2:1,每浓度3个柱体积)梯度洗脱后,合并第16-29流份(F3-6-2),经凝胶柱层析(二氯甲烷:甲醇1:1系统)分离后,通过HPLC(55%甲醇:水系统)等度洗脱,得到单体P1(3mg)。第4份(F.4,0.391g)经硅胶柱层析,二氯甲烷:甲醇系统(30:1、20:1、10:1、8:1、5:1、3:1、2:1,每浓度5个柱体积;V/V)梯度洗脱分离得到66流份(图1-2)。合并9-17流份(Fr.4-3),经凝胶柱(甲醇系统)层析分得37流份,合并34-37流份得到单体P2(47mg)。分离过程如图13。(3.2)乙酸乙酯:甲醇(10:1)部位共105g,溶解后硅胶(105g,100-200目)拌样,经硅胶(1000g,100-200目)进行柱层析,以二氯甲烷:甲醇系统(1:0、100:1、50:1、30:1、10:1、8:1、5:1、3:1、2:1;V/V),梯度洗脱共收集141流份,每500ml旋出作为1流份,经薄层色谱点板后分析合并为6份,第1份(Fr.1-5),第2份(Fr.6),第3份(Fr.7-46),第4份(Fr.47-79),第5份(Fr.80-101),第6份(Fr.102-141)。将合并后的F1-F6各部分进行DPPH清除活性测定(表6),各部位DPPH自由基相对清除活性顺序为:F6>F4>F5>F3>F2>F1。表6乙酸乙酯:甲醇(10:1)部位各部分DPPH自由基清除率选取乙酸乙酯:甲醇(10:1)部位的第6份(F6)进行进一步分离。第6份(F6)经凝胶柱除去色素,已除去色素部位(F6-2,23.7g)溶解后硅胶(27g,100-200目)拌样,硅胶(200-300目)装柱,用二氯甲烷:甲醇(体积比20:1,10:1,5:1,3:1,2:1)梯度洗脱,共收集95流份。根据薄层色谱点板结果合并为5份,将合并后的F6-2-1至F6-2-5各部分进行DPPH清除活性测定(表7),DPPH相对清除活性为:F6-2-2>F6-2-5>F6-2-3,F6-2-4>F6-2-1。其中第5份(二氯甲烷:甲醇体积比2:1,记为F6-2-5)重结晶得化合物P6。表7乙酸乙酯:甲醇(10:1)部位F6-2分离各部分DPPH自由基清除率部位IC50mg/mlF6-2-10.461±0.018F6-2-20.070±0.001aF6-2-30.130±0.003cF6-2-40.135±0.007cF6-2-50.080±0.005bP值<0.001其中第2份(二氯甲烷:甲醇体积比10:1,记为F6-2-2,合并第24-36流份,0.258g)经ODS柱按30%-100%甲醇:水梯度洗脱,得到63流份,根据薄层色谱结果合并为18份,其中第24-27流份合并经HPLC(30min,40%-90%甲醇:水系统流动相,19ml/min)制备得到P5(如图14)。表8车前分离单体化合物各单体物质的波谱数据及结构判定如下:P1:甘草素无色针晶(丙酮),ESI-MS:m/z257[M+H]+;1H-NMR(500MHz,CD3OCD3)δ:7.72(1H,d,J=8.5Hz,H-5),7.40(2H,d,J=8.5Hz,H-2′,6′),6.90(2H,d,J=8.5Hz,H-3′,5′),6.57(1H,dd,J=8.5,2.0Hz,H-6),6.42(1H,d,J=2.0Hz,H-8),5.44(1H,dd,J=13.0,3.0Hz,H-2),3.04(1H,dd,J=16.5,13.0Hz,H-3a),2.67(1H,dd,J=16.5,3.0Hz,H-3b);13C-NMR(125MHz,CD3OCD3)δ:44.7(C-3),80.6(C-2),103.7(C-8),111.3(C-6),115.1(C-10),116.1(C-3′,5′),128.9(C-2′,6′),129.5(C-5),131.3(C-1′),158.6(C-4′),164.6(C-9),165.5(C-7),190.5(C-4)。以上数据证明为如下结构的甘草素。P2:芹菜素淡黄色粉末状结晶(甲醇),mp.347-348℃。ESI-MSm/z:271[M+H]+;1H-NMR(500MHz,DMSO-d6)δ:12.96(1H,s,5-OH),7.92(2H,d,J=9.0Hz,H-2′,6′),6.92(2H,d,J=9.0Hz,H-3′,5′),6.78(1H,s,H-3),6.48(1H,d,J=2.0Hz,H-8),6.19(1H,d,J=2.0Hz,H-6)。以上数据鉴定为如下结构的芹菜素(apigenin)。P6:七叶内酯黄色针晶(甲醇),mp.268-270℃。ESI-MSm/z:179[M+H]+;1H-NMR(500MHz,DMSO-d6)δ:7.85(1H,d,J=9.5Hz,H-4),6.15(1H,d,J=9.5Hz,H-3),6.97(1H,s,H-5),6.74(1H,s,H-8),10.20(1H,s,OH),9.39(1H,s,OH)。由以上数据确定该化合物为如下结构的七叶内酯(aesculetin)。P5:4,5-二咖啡酰基奎宁酸甲酯淡黄色粉末,ESI-MSm/z:531[M+H]+;1H-NMR(500MHz,DMSO-d6)δ:7.04(1H,d,J=1.5Hz,H-2′),7.02(1H,d,J=1.5Hz,H-2″),6.99(2H,m,H-6′,6″),6.76(2H,d,J=8.0Hz,H-5′,5″),7.52(1H,d,J=16.0Hz,H-7′),6.28(1H,d,J=16.0Hz,H-8′),7.42(1H,d,J=16.0Hz,H-7″),6.14(1H,d,J=16.0Hz,H-8″),5.26(1H,m,H-3),4.96(1H,dd,J=6.5,3.5Hz,H-4),4.13(1H,m,H-5),3.61(3H,s,OCH3),2.15(3H,m,H-2,6b),1.93(1H,m,H-2,6a);13C-NMR(125MHz,DMSO-d6)δ:72.5(C-1),34.9(C-2),66.6(C-3),70.1(C-4),70.0(C-5),34.5(C-6),175.1(C-7),125.5(C-1′),125.1(C-1″),114.8(C-2′),114.6(C-2″),145.7(C-3′),145.2(C-3″),149.1(C-4′),149.0(C-4″),115.8(C-5′),116.0(C-5″),121.6(C-6′),121.4(C-6″),146.0(C-7′,7″),113.2(C-8′),114.2(C-8″),106.1(C-9′),165.4(C-9″),52.0(OCH3)。以上数据判定为4,5-二咖啡酰基奎宁酸甲酯(4,5-Dicaffeoylquinicacidmethylester),结构如下:实施例4车前优化工艺提取物质所鉴定化合物的抗氧化活性将分离得到的化合物通过H2O2构造Caco2损伤模型验证其抗氧化活性,结果如下:4.1七叶内酯缓解过氧化氢损伤作用300μg/ml的七叶内酯预孵育显著缓解了1000μMH2O2刺激导致的细胞活力下降,与空白对照无显著性差异,但随着加入七叶内酯浓度的增加缓解作用逐步加强(表9)。表9P6(七叶内酯)对1000μMH2O2刺激下细胞活率的影响(保护作用)通过不同浓度七叶内酯预处理再进行H2O2刺激,100μg/ml及以上浓度的七叶内酯显著增强了细胞的SOD、CAT和GCS的转录,并随着七叶内酯浓度提高转录进一步增强。低浓度七叶内酯预处理减弱了Nrf2转录,而在高浓度下进一步显著增加了Nrf2的转录(表10)。表10P6(七叶内酯)对1000μMH2O2刺激下抗氧化相关基因转录水平的影响(保护作用)4.2芹菜素缓解过氧化氢损伤作用1000μMH2O2较空白对照显著降低了细胞活力,芹菜素预处理显著并剂量依赖性的改善了过氧化氢引起的细胞活力降低(表11)。表11P2(芹菜素)对1000μMH2O2刺激下细胞活率的影响(保护作用)不同浓度芹菜素预处理后进行1000μMH2O2损伤,进一步降低了Nrf2和CAT的转录水平。但低浓度芹菜素(31.25μg/ml)预处理进一步提高了GCS的转录,而高浓度芹菜素预处理显著降低损伤后GCS转录并存在剂量效应。高浓度的芹菜素同时可提高损伤后SOD的转录(表12)。表12P2(芹菜素)对1000μMH2O2刺激下抗氧化相关基因转录水平的影响(保护作用)以上的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通工程技术人员对本发明的技术方案作出的各种变型和改进,均应落入本发明的权利要求书确定的保护范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1