一种绿藻多糖及其制备方法与流程
本发明涉及海洋生物多糖技术领域,具体来说涉及一种绿藻多糖及其制备方法。
背景技术:
海藻自古就在我国沿海民间直接用作药材医治各种疾病,有关海藻中有效成分的分离及其化学组成的研究,始于上世纪50年代中期。至今,从海藻中分离出的各种化合物中,多糖类化合物占有重要地位。多糖作为海藻细胞结构的主要支撑物质,大量存在于其细胞壁和细胞间质中。由于海藻多糖独特的结构和性质,引起人们的广泛关注。有关海藻多糖的化学组成、结构和生物活性已被大量研究。但是,这些研究主要集中于来自于各种褐藻和红藻的多糖,而有关绿藻中的多糖的研究极少。
目前,绿藻多糖的研究主要集中于绿藻中的石莼属、松藻属、浒苔属、礁膜属、蕨藻属和刚毛藻属中的藻类,其化学组成和结构随着绿藻种类的不同而不同,已有资料表明这些多糖主要分为两类:木糖-阿拉伯糖-半乳糖聚合物,这类多糖以阿拉伯糖和半乳糖为主要组分,并含有较高的硫酸根,其代表藻类为松藻、蕨藻和刚毛藻等;葡萄糖醛酸-木糖-鼠李糖聚合物,此类多糖以鼠李糖、木糖和葡萄糖醛酸为主要组分,代表藻属为石莼、礁膜、浒苔和顶管藻等。由于绿藻多糖结构的复杂性和不均一性,大大限制了对其深入研究。迄今尚未见有关从绿藻中提取分离到由葡萄糖醛酸和鼠李糖为组成单元、且葡萄糖醛酸位于支链的硫酸多糖的报道。
技术实现要素:
本发明的目的在于提供一种绿藻多糖及其制备方法,本发明的绿藻多糖是以α-l-(1,3)-连接的鼠李糖和α-l-(1,2)-连接的鼠李糖为主链,β-d-葡萄糖醛酸位于支链的一种结构新颖的海洋硫酸多糖。本发明的绿藻多糖具有明显抗凝血活性、纤溶和溶栓活性以及降血糖降血脂活性,可用于制备抗凝血和降血糖药物及保健食品。
为此,本发明提供了一种绿藻多糖,所述绿藻多糖是由鼠李糖和葡萄糖醛酸组成的一种硫酸多糖,其中,鼠李糖的摩尔百分比为96.12%,葡萄糖醛酸的摩尔百分比为3.88%;所述绿藻多糖的主链由α-l-(1,3)-连接的鼠李糖和α-l-(1,2)-连接的鼠李糖组成,α-l-(1,3)-连接的鼠李糖和α-l-(1,2)-连接的鼠李糖的摩尔比为5:3,在α-l-(1,3)-连接的鼠李糖的c2位存在支链;所述绿藻多糖的支链由α-l-(1,3)-连接的鼠李糖和末端连接的β-d-葡萄糖醛酸组成,β-d-葡萄糖醛酸位于支链的非还原端,β-d-葡萄糖醛酸与鼠李糖之间以1,3糖苷键连接;所述绿藻多糖的结构分子式如式ⅰ所示,其中,n=30,m=0-5,r1=h或so3h,r2=h或so3h,r3=h或so3h,r4=h或so3h;
本发明还提供了绿藻多糖的制备方法,所述方法包括如下步骤:
(1)向干燥的袋礁膜中加水浸泡形成匀浆,在20℃-24℃下提取0.5-4小时,分离出清液;其中,水的质量为袋礁膜质量的10-60倍;
(2)将所得清液减压浓缩、脱盐得到第一多糖溶液,将所述第一多糖溶液减压浓缩、干燥得到多糖,将所述多糖加水溶解得到第二多糖溶液,所述第二多糖溶液中所述多糖的质量分数为0.1%-6%;
(3)将所述第二多糖溶液通过离子交换色谱柱分离和凝胶色谱柱分离,然后减压浓缩、脱盐,将脱盐后的溶液减压浓缩、干燥,得到所述绿藻多糖。
与现有技术相比,本发明的优点和积极效果是:本发明提供了一种绿藻多糖及其制备方法,本发明的绿藻多糖是以α-l-(1,3)-连接的鼠李糖和α-l-(1,2)-连接的鼠李糖为主链,β-d-葡萄糖醛酸位于支链的一种结构新颖的海洋硫酸多糖。本发明的绿藻多糖具有明显抗凝血活性、纤溶和溶栓活性以及降血糖降血脂活性,可用于制备抗凝血和降血糖药物及保健食品。本发明的绿藻多糖来源于天然海藻,对人体无毒副作用,可以通过添加各种附剂制成片剂、散剂、颗粒剂、胶囊等作为预防和治疗血栓栓塞性疾病的抗凝血药物以及预防和治疗糖尿病的降血糖药物,将具有良好的市场应用前景。
结合附图阅读本发明的具体实施方式后,本发明的其他特点和优点将变得更加清楚。
附图说明
图1是所述绿藻多糖的纯度测定的高效凝胶渗透色谱和葡聚糖标准品的分子量对数对保留时间(高效凝胶渗透色谱法测定)绘制的标准曲线及其回归方程;
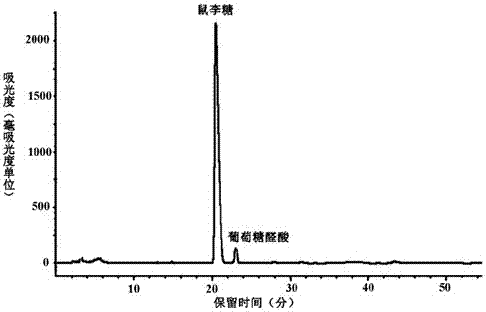
图2是本发明所述绿藻多糖的单糖组成测定的柱前衍生高效液相色谱;
图3是本发明所述绿藻多糖中单糖的绝对构型测定的柱前衍生高效液相色谱;
图4是本发明所述绿藻多糖的红外光谱;
图5是本发明所述绿藻多糖脱硫后的1hnmr谱;
图6是本发明所述绿藻多糖脱硫后的13cnmr谱;
图7是本发明所述绿藻多糖脱硫后的1h-1hcosy谱;
图8是本发明所述绿藻多糖脱硫后的1h-13chsqc谱;
图9是本发明所述绿藻多糖脱硫后的1h-13chmbc谱;
图10是本发明所述绿藻多糖的1hnmr谱;
图11是本发明所述绿藻多糖的13cnmr谱;
图12是本发明所述绿藻糖的1h-1hcosy谱;
图13是本发明所述绿藻多糖的1h-13chsqc谱;
图14是本发明所述绿藻多糖的1h-1hnoesy谱;
图15是本发明所述绿藻多糖经降解得到的寡糖在凝胶色谱柱bio-gelp4的分离纯化图;
图16是本发明所述绿藻多糖经降解得到的寡糖组分s1的esi-ms谱;
图17是本发明所述绿藻多糖经降解得到的寡糖组分s2的esi-ms谱;
图18是本发明所述绿藻多糖经降解得到的寡糖组分s3的esi-ms谱;
图19是本发明所述绿藻多糖经降解得到的寡糖组分s4的esi-ms谱;
图20是本发明所述绿藻多糖经降解得到的寡糖组分s1的碎片离子m/z243的esi-cid-ms/ms谱和质谱断裂模式示意图;
图21是本发明所述绿藻多糖经降解得到的寡糖组分s2的碎片离子m/z389的esi-cid-ms/ms谱和质谱断裂模式示意图;
图22是本发明所述绿藻多糖经降解得到的寡糖组分s2的碎片离子m/z339的esi-cid-ms/ms谱和质谱断裂模式示意图;
图23是本发明所述绿藻多糖经降解得到的寡糖组分s3的碎片离子m/z485的esi-cid-ms/ms谱和质谱断裂模式示意图;
图24是本发明所述绿藻多糖经降解得到的寡糖组分s4的碎片离子m/z419的esi-cid-ms/ms谱和质谱断裂模式示意图;
图25是本发明所述绿藻多糖经降解得到的寡糖组分s4的碎片离子m/z565的esi-cid-ms/ms谱和质谱断裂模式示意图;
图26是本发明所述绿藻多糖对大鼠血浆纤维蛋白(原)降解产物含量的影响;统计学意义:**p<0.01,与空白组比较;##p<0.01,与尿激酶组比较;
图27是本发明所述绿藻多糖对大鼠血浆d-二聚体含量的影响;统计学意义:**p<0.01,与空白组比较;##p<0.01,与尿激酶组比较;
图28是本发明所述绿藻多糖对大鼠血浆纤溶酶原激活抑制物水平的影响;统计学意义:*p<0.05,**p<0.01,与空白组比较;##p<0.01,与尿激酶组比较;
图29是本发明所述绿藻多糖的体外血凝块溶解率分析结果;统计学意义:**p<0.01,与空白组比较;#p<0.05,##p<0.01,与尿激酶组比较;
图30是本发明所述绿藻多糖对hepg2细胞葡萄糖消耗能力的影响;统计学意义:**p<0.01,与空白组比较;
图31是本发明所述绿藻多糖对棕榈酸诱导的胰岛素抵抗hepg2细胞葡萄糖消耗能力的影响;统计学意义:*p<0.05,与空白组比较;#p<0.05,与模型组比较;
图32是本发明所述绿藻多糖对棕榈酸诱导的胰岛素抵抗hepg2细胞甘油三酯含量的影响;统计学意义:**p<0.01,与空白组比较;##p<0.01,与模型组比较;
图33是本发明所述绿藻多糖对棕榈酸诱导的胰岛素抵抗hepg2细胞总胆固醇含量的影响;统计学意义:**p<0.01,与空白组比较;##p<0.01,与模型组比较。
具体实施方式
以下对本发明的具体实施方式进行详细说明,应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
本发明的绿藻多糖是由鼠李糖和葡萄糖醛酸组成的一种硫酸多糖,其中,鼠李糖的摩尔百分比为96.12%,葡萄糖醛酸的摩尔百分比为3.88%;所述绿藻多糖的主链由α-l-(1,3)-连接的鼠李糖和α-l-(1,2)-连接的鼠李糖组成,α-l-(1,3)-连接的鼠李糖和α-l-(1,2)-连接的鼠李糖的摩尔比为5:3,在α-l-(1,3)-连接的鼠李糖的c2位存在支链;所述绿藻多糖的支链由α-l-(1,3)-连接的鼠李糖和末端连接的β-d-葡萄糖醛酸组成,β-d-葡萄糖醛酸位于支链的非还原端,β-d-葡萄糖醛酸与鼠李糖之间以1,3糖苷键连接;所述绿藻多糖的结构分子式如式ⅰ所示,其中,n=30,m=0-5,r1=h或so3h,r2=h或so3h,r3=h或so3h,r4=h或so3h;
本发明的绿藻多糖是以α-l-(1,3)-连接的鼠李糖和α-l-(1,2)-连接的鼠李糖为主链,β-d-葡萄糖醛酸位于支链的一种结构新颖的海洋硫酸多糖。本发明的绿藻多糖具有明显抗凝血活性、纤溶和溶栓活性以及降血糖降血脂活性,可用于制备抗凝血和降血糖药物及保健食品。本发明的绿藻多糖来源于天然海藻,对人体无毒副作用,可以通过添加各种附剂制成片剂、散剂、颗粒剂、胶囊等作为预防和治疗血栓栓塞性疾病的抗凝血药物以及预防和治疗糖尿病的降血糖药物,将具有良好的市场应用前景。
绿藻多糖的平均分子量为58.4千道尔顿。
在绿藻多糖的鼠李糖中,17.77%的α-l-(1,3)-连接的鼠李糖被硫酸化形成硫酸基并位于α-l-(1,3)-连接的鼠李糖的c2位,4.15%的α-l-(1,2)-连接的鼠李糖被硫酸化形成硫酸基并位于α-l-(1,2)-连接的鼠李糖的c3位。
在绿藻多糖的糖链中,每10个鼠李糖基中存在一个支链。
本发明还提供了绿藻多糖的制备方法,所述方法包括如下步骤:
(1)向干燥的袋礁膜中加水浸泡形成匀浆,在20℃-24℃下提取0.5-4小时,分离出清液;其中,水的质量为袋礁膜质量的10-60倍;
(2)将所得清液减压浓缩、脱盐得到第一多糖溶液,将所述第一多糖溶液减压浓缩、干燥得到多糖,将所述多糖加水溶解得到第二多糖溶液,所述第二多糖溶液中所述多糖的质量分数为0.1%-6%;
(3)将所述第二多糖溶液通过离子交换色谱柱分离和凝胶色谱柱分离,然后减压浓缩、脱盐,将脱盐后的溶液减压浓缩、干燥,得到所述绿藻多糖。
步骤(1)中,向干燥的袋礁膜中加水浸泡形成匀浆的方法为:将干燥的袋礁膜直接浸泡于水中形成匀浆,或者将干燥的袋礁膜研磨或绞碎后与水混合形成匀浆,或者将干燥的袋礁膜加工成干燥粉末与水混合形成匀浆。
步骤(2)和步骤(3)中,脱盐的方法为透析脱盐。
步骤(2)和步骤(3)中,干燥的方法为冷冻干燥,或者向减压浓缩后的多糖溶液中加入1-6倍多糖溶液体积的、质量分数为95%乙醇使多糖沉淀,将所得沉淀在40℃-80℃烘箱中干燥0.1-6小时。
步骤(3)中,离子交换色谱柱为色谱柱qsepharosefastflow,依次用蒸馏水、0.5mol/l氯化钠水溶液、1.0mol/l氯化钠水溶液、1.5mol/l氯化钠水溶液和2.0mol/l氯化钠水溶液洗脱。
步骤(3)中,凝胶色谱柱为色谱柱sephacryls-400hr,所用的洗脱液为0.2mol/l碳酸氢铵水溶液。
实施例1
(1)向50克干燥的袋礁膜中加1500克水浸泡形成匀浆,在21℃下搅拌提取3.0小时,离心弃去沉淀,分离出清液;
(2)将所得清液减压浓缩、透析脱盐得到第一多糖溶液,透析脱盐的步骤包括:将减压浓缩后的清液置于截留分子量为3500道尔顿的透析袋中透析3天,电导率仪测试透析后外液至恒定值时停止透析;
将第一多糖溶液减压浓缩、向浓缩后第一多糖溶液中加入质量分数为95%乙醇使多糖沉淀,乙醇的体积为第一多糖溶液体积的四倍,将所得沉淀在40℃烘箱中干燥4小时,得到5克多糖。将多糖加水溶解得到第二多糖溶液,每毫升第二多糖溶液中多糖的质量为50毫克。
(3)将第二多糖溶液通过阴离子交换柱qsepharosefastflow分离,依次分别用蒸馏水、0.5mol/l氯化钠水溶液、1.0mol/l氯化钠水溶液、1.5mol/l氯化钠水溶液和2.0mol/l氯化钠水溶液洗脱2个柱体积,收集2.0mol/l氯化钠水溶液的洗脱液,将所得多糖溶液减压浓缩,然后进行透析去除盐,将除去盐后的多糖溶液减压浓缩,干燥后加蒸馏水溶解,使溶液中多糖的质量分数为5%;
再通过凝胶色谱柱sephacryls-400/hr进一步分离,用0.2mol/l碳酸氢铵水溶液洗脱,硫酸-苯酚法检测,收集多糖洗脱峰的洗脱液,将所得多糖溶液减压浓缩,然后进行透析去除盐,将除去盐后的多糖溶液减压浓缩,冷冻干燥,得到0.14克绿藻多糖。
以下为绿藻多糖的结构测试和分析,包括绿藻多糖的单糖组成分析、甲基化分析、核磁共振波谱、寡糖的电喷雾负离子各离子峰的表征和电喷雾电离-碰撞诱导解离二级质谱的序列分析等。
绿藻多糖为白色粉末,其硫酸基重量百分比为28.83%(氯化钡-明胶比浊法测定),其糖醛酸重量百分比为5.09%(硫酸咔唑法)。
绿藻多糖的纯度和分子量通过高效凝胶渗透色谱法测定,结果如图1左侧的视图a所示,绿藻多糖呈现单一对称峰,说明绿藻多糖的纯度高。在同样的分析条件下,绘制葡聚糖标准品(分子量为:708,344,200,107,47.1和21.1千道尔顿)的分子量对数对保留时间的标准曲线,并获得标准曲线的回归方程,结果如图1右侧的视图b所示;通过将绿藻多糖的保留时间(如图1左侧的视图a所示)代入图1右侧的视图b中的标准曲线回归方程,计算得到绿藻多糖的平均分子量为58.4千道尔顿。
绿藻多糖的单糖组成通过柱前衍生高效液相色谱法测定,结果如图2所示。根据保留时间和峰面积进行计算,并与鼠李糖、葡萄糖、葡萄糖胺、岩藻糖、木糖、阿拉伯糖、甘露糖、半乳糖、葡萄糖醛酸和半乳糖醛酸进行对照,可知绿藻多糖主要含有鼠李糖及少量葡萄糖醛酸,鼠李糖的摩尔百分比为96.12%,葡萄糖醛酸的摩尔百分比为3.88%。
绿藻多糖中单糖的绝对构型通过柱前衍生高效液相色谱法测定,结果如图3所示。结果表明绿藻多糖中鼠李糖的保留时间为29.88分钟,其与l-鼠李糖标准品的保留时间一致,说明绿藻多糖中的鼠李糖的绝对构型为l型,葡萄糖醛酸的保留时间为18.51分钟,其与d-葡萄糖醛酸标准品的保留时间一致,说明绿藻多糖中的葡萄糖醛酸的绝对构型为d型。
绿藻多糖的红外光谱如图4所示,波数3458cm–1的强吸收峰为o-h的伸缩振动,2928cm–1吸收峰为c-h的伸缩振动吸收,1640cm–1和1429cm–1吸收峰为coo–的非对称以及对称伸缩振动峰,1240cm–1吸收峰为硫酸基的s=o伸缩振动,1051cm–1吸收峰为c-o-c环内醚的c-o伸缩振动和o–h的变角振动,850cm–1吸收峰为硫酸基的c-o-s的伸缩振动吸收。
绿藻多糖中糖基的连接方式和硫酸基位置首先通过甲基化分析测定,绿藻多糖脱硫前和脱硫后的甲基化分析结果表明,脱硫前绿藻多糖主要含有1,3、1,2和1,2,3连接的l-鼠李糖,三者的摩尔百分比约为41.08:25.69:33.23;脱硫后与脱硫前的绿藻多糖相比,1,2连接的鼠李糖和1,3连接的鼠李糖的摩尔百分比增大,1,2,3连接的鼠李糖的摩尔百分比减小,脱硫后绿藻多糖中1,3、1,2和1,2,3连接的鼠李糖的摩尔百分比约为58.85:29.84:11.31。说明硫酸基位于1,2连接的鼠李糖的c3位和1,3连接的鼠李糖的c2位,而且,可以推断绿藻多糖中鼠李糖总数的21.92%(摩尔百分比)被硫酸基取代,且1,3连接的鼠李糖的17.77%被硫酸基取代,1,2连接的鼠李糖的4.15%被硫酸基取代。另外,通过对绿藻多糖中的糖醛酸还原后产物的甲基化分析说明,绿藻多糖中的葡萄糖醛酸是以末端连接的形式存在。
图5为脱硫后的绿藻多糖的核磁共振氢谱(1hnmr谱),化学位移5.00~5.50ppm,为α吡喃型鼠李糖端基氢信号所在的区域(以下将α吡喃型鼠李糖基简写为α-rhap),异头区显示5个异头氢信号,分别为5.00、5.07、5.23、5.29和5.38ppm,其摩尔比为1.00:1.03:1.01:0.21:0.21,依次命名为a、b、c、d和e。高场区1.35ppm为鼠李糖基的c6甲基氢信号,3.30~4.50ppm为鼠李糖糖环碳上无取代基团时h2~h5的信号。
图6为脱硫后的绿藻多糖的核磁共振碳谱(13cnmr谱),图6中出现三个主要的异头碳信号:103.54、102.41和102.22ppm,鼠李糖基的c2~c5信号主要集中于70~80ppm,18.26ppm为鼠李糖基的c6信号,由3.89ppm的h5信号和70.80ppm的c5信号也可以推断出鼠李糖基为α型。由于葡萄糖醛酸含量较低且信号重叠严重,在核磁上确定它的信号非常困难。13cnmr谱中105.72ppm为非还原端葡萄糖醛酸基的异头碳信号,177.33ppm为葡萄糖醛酸的羧基碳信号。
图7为脱硫后的绿藻多糖的同核化学位移相关谱(1h-1hcosy谱),图8为脱硫后的绿藻多糖的异核单量子化学位移相关谱(1h-13chsqc谱)。从图7和图8可以得出,a和b糖基中,5.00和5.07ppm的h1与103.54ppm的c1相关,由于79.43ppm的c3位化学位移向低场移动,因此a和b糖基归属为→3)-α-l-rhap-(1→。c糖基5.23ppm的h1与102.41ppm的c1相关,且其相关信号4.11/79.01ppm的h2/c2存在,表明c糖基为→2)-α-l-rhap-(1→。d糖基5.29ppm的h1与102.22ppm的c1相关,其相关信号4.11/79.01ppm的h2/c2和4.00/79.43ppm的h3/c3存在,证明了c2和c3取代,因此d糖基为→2,3)-α-l-rhap-(1→。e糖基归属为→2)-α-l-rhap(3so4)-(1→,其5.38ppm的h1与102.22ppm的c1相关,由于4.61ppm的h3被硫酸基取代,因此,化学位移向低场移动。e糖基的出现表明多糖的脱硫并不完全,但是由于其信号强度较弱,因此大部分硫酸基已经被脱除。
图9为脱硫后的绿藻多糖的异核多键化学位移相关谱(1h-13chmbc谱),图9给出不同糖环上碳氢之间的远程相关信号。a糖基的h1与c、d和e糖基的c2相关,推出片段→3)-α-l-rhap-(1→2)-α-l-rhap→、→3)-α-l-rhap-(1→2,3)-α-l-rhap→和→3)-α-l-rhap-(1→2)-α-l-rhap(3so4)-(1→。b糖基的h1与a和d糖基的c3相关,推出→3)-α-l-rhap-(1→3)-α-l-rhap→和→3)-α-l-rhap-(1→2,3)-α-l-rhap→。c糖基的h1与a和b糖基的c3相关,可推出→2)-α-l-rhap-(1→3)-α-l-rhap→。此外,相关信号h1(d)/c2(c)、h1(d)/c2(e)、h1(e)/c3(a)和h1(e)/c3(b)表明可能存在以下序列片段→2,3)-α-l-rhap-(1→2)-α-l-rhap→、→2,3)-α-l-rhap-(1→2)-α-l-rhap(3so4)→和→2)-α-l-rhap(3so4)-(1→3)-α-l-rhap→。
图10为绿藻多糖的核磁共振氢谱(1hnmr谱),存在六种异头氢信号,化学位移分别为5.00、5.07、5.23、5.29、5.38和5.50ppm,均为α吡喃型的鼠李糖端基氢信号,依次命名为a、b、c、d、e和f,其摩尔比为1.00:2.40:2.07:0.50:0.88:1.45。与脱硫后的绿藻多糖的1hnmr谱相比,绿藻多糖的1hnmr谱显示了一个额外的异头氢信号5.50ppm,而且5.38和5.50ppm的异头氢信号强度较高。图11为绿藻多糖的核磁共振碳谱(13cnmr谱),图12为绿藻多糖的同核化学位移相关谱(1h-1hcosy谱),图13为绿藻多糖的异核单量子化学位移相关谱(1h-13chsqc谱),图14为绿藻多糖的核欧沃豪斯效应相关谱(1h-1hnoesy谱);通过图10-图13可知,f糖基归属为→3)-α-l-rhap(2so4)-(1→,其h1与100.61ppm的c1相关;e糖基归属为→2)-α-l-rhap(3so4)-(1→,其h1与100.61ppm的c1相关;根据绿藻多糖的脱硫后多糖的信号归属,a和b糖基的h1信号与103.16和102.78ppm的c1信号相关,h3(3.92/3.96ppm)与c3(78.30ppm)相关,因此a和b归属为→3)-α-l-rhap-(1→;c糖基的h1与101.42ppm的c1相关,4.30ppm的h2与78.86ppm的c-2相关,c归属为→2)-α-l-rhap-(1→;d糖基归属为→2,3)-α-l-rhap-(1→,其h1与101.42ppm的c1相关,h2/c2和h3/c3的相关信号分别为4.30/78.86ppm和3.97/78.30ppm。
从图14可以看出不同糖环上氢氢之间的远程相关信号,a糖基的h1与4.30ppm的c糖基、d糖基和e糖基的h2相关,表明存在→3)-α-l-rhap-(1→2)-α-l-rhap-(1→、→3)-α-l-rhap-(1→2,3)-α-l-rhap-(1→和→3)-α-l-rhap-(1→2)-α-l-rhap(3so4)-(1→片段。b糖基的h1与a糖基、f糖基的h3(3.92/4.11ppm)相关,此外,b糖基的3.96ppm的h3与f糖基的5.50ppm的h1相关,表明存在以下片段→3)-α-l-rhap-(1→3)-α-l-rhap-(1→、→3)-α-l-rhap-(1→3)-α-l-rhap(2so4)-(1→和→3)-α-l-rhap(2so4)-(1→3)-α-l-rhap-(1→。c糖基、d糖基和e糖基的h1(5.23、5.29、5.38ppm)与b糖基的h3(3.96ppm)相关,表明存在以下片段→2)-α-l-rhap-(1→3)-α-l-rhap-(1→、→2,3)-α-l-rhap-(1→3)-α-l-rhap-(1→和→2)-α-l-rhap(3so4)-(1→3)-α-l-rhap-(1→。c糖基的h1与f糖基的h3相关,表明存在→2)-α-l-rhap-(1→3)-α-l-rhap(2so4)-(1→。
为进一步研究绿藻多糖的结构,采用酸解法对绿藻多糖进行降解,选用凝胶柱bio-gelp4对降解产物进行分离纯化。对绿藻多糖经降解所得的寡糖片段采用电喷雾电离质谱(esi-ms)测定寡糖片段的分子量,采用电喷雾电离-碰撞诱导解离二级质谱(esi-cid-ms/ms)技术对寡糖组分的结构进行研究,最终确定绿藻多糖的结构。
绿藻多糖的降解:绿藻多糖加水配成质量百分比为1%的绿藻多糖溶液,加入适量盐酸至反应体系盐酸浓度为0.1mol/l,在60℃时反应9小时,所得水解物用1.0mol/l氢氧化钠中和,然后置于截留分子量为100道尔顿的透析袋中透析3天,将所得水解物溶液在40℃减压浓缩,然后向浓缩液中加入质量分数为95%的乙醇(浓缩液与乙醇的体积比3:1),通过离心(4500转/分钟,10分钟),获得了两种产物,即上清液和沉淀。上清液部分经减压浓缩后冻干,沉淀部分用无水乙醇脱水后于40℃烘干。通过柱前衍生高效液相色谱测定它们的单糖组成,结果表明,所得沉淀部分中仅含有鼠李糖,而冻干后的上清液部分含有鼠李糖和葡萄糖醛酸。由此得知,葡萄糖醛酸片段更容易被降解下来,推测其位于绿藻多糖的支链。
寡糖的分离:采用凝胶柱bio-gelp4(100×1.6cm)层析柱对绿藻多糖的降解产物,即冻干后的上清液部分进行分离,洗脱液为0.2mol/l碳酸氢铵水溶液,采用苯酚硫酸法检测,收集主峰部分,脱盐后冻干即得寡糖,所得寡糖依次命名为s1、s2、s3和s4。图15是绿藻多糖经降解得到的寡糖在凝胶色谱柱bio-gelp4的分离纯化图,通过负离子方式的esi-ms谱分析各寡糖组分的分子量,结果如图16-图19所示,从图16-图19可以看出,s1为硫酸化鼠李单糖,s2主要为硫酸化鼠李二糖,s3为硫酸化或非硫酸化二糖与三糖,s4为高聚合度的寡糖。葡萄糖醛酸片段主要存在于寡糖组分s2、s3和s4中,以鼠李单糖和糖醛酸(r+ga),鼠李二糖和糖醛酸(r2+ga),硫酸化鼠李单糖和糖醛酸(rs+ga)以及硫酸化鼠李二糖和糖醛酸(r2s+ga)的形式存在,而硫酸化鼠李单糖(rs)和硫酸化鼠李二糖(r2s)是寡糖s1与s2的主要组分。r代表鼠李糖,ga代表葡萄糖醛酸,s代表硫酸基。
寡糖的序列分析:应用电喷雾电离质谱并利用撞击分散诱导技术对所得的寡糖进行序列分析。首先,对寡糖组分s1中硫酸化鼠李单糖进行序列分析,寡糖组分s1的碎片离子质荷比(以下将质荷比简写为m/z)243的esi-cid-ms/ms谱如图20中左侧的视图a所示。从硫酸化鼠李单糖m/z243的二级质谱图中可以看到,主要有m/z139环内断裂碎片离子,归属为0,2x,因此硫酸基位于c2位。少量m/z225为硫酸化鼠李单糖(rs)失去一分子水所产生的离子。寡糖组分s1的碎片离子m/z243的质谱断裂模式示意图如图20中右侧的视图b所示。
对寡糖组分s2中硫酸化鼠李二糖(r2s)以及葡萄糖醛酸鼠李二糖(r+ga)进行序列分析。寡糖组分s2的碎片离子m/z389的esi-cid-ms/ms谱如图21左侧的视图a所示。在硫酸化鼠李二糖m/z389的二级质谱中,碎片离子m/z225和m/z243由鼠李糖糖环间的断裂产生,分别归属为b1和c1。由此推断,硫酸化鼠李二糖是在硫酸化鼠李单糖的基础上连接了另一个鼠李糖。其他碎片离子峰均由鼠李糖环内断裂产生,m/z285和m/z315分别归属为0,2x1和0,3x1。由上述分析可以推断得出,硫酸化鼠李二糖的结构为α-l-rhap(2so4)-(1→3)-α-l-rhap。寡糖组分s2的碎片离子m/z389的的质谱断裂模式示意图如图21右侧的视图b所示。
寡糖组分s2的碎片离子m/z339的esi-cid-ms/ms谱如图22左侧的视图a所示,在葡萄糖醛酸鼠李糖m/z339的二级质谱中,碎片离子m/z321由r+ga脱去一分子水产生。在m/z321的三级质谱中,m/z193仍旧出现,而且只在还原端葡萄糖醛酸中出现的2,5a2–型离子并没有出现,因此葡萄糖醛酸位于非还原端。葡萄糖醛酸鼠李二糖(r+ga)的结构为β-d-glcpa-(1→3)-α-l-rhap。寡糖组分s2的碎片离子m/z339的质谱断裂模式示意图如图22右侧的视图b所示。
对寡糖组分s3的碎片离子m/z485的esi-cid-ms/ms谱如图23左侧的视图a所示,葡萄糖醛酸鼠李三糖m/z485的二级质谱显示了与葡萄糖醛酸鼠李二糖m/z339相似的碎片离子,给出了鼠李糖环间断裂离子c3(m/z467)、c2(m/z339)、b2(m/z321)、c1(m/z193)以及环内断裂离子峰1,3a2(m/z235),由此推断葡萄糖醛酸鼠李三糖是在葡萄糖醛酸鼠李二糖的基础上又连接了一个鼠李糖,其结构为β-d-glca-(1→3)-α-l-rhap-(1→3)-α-l-rhap。根据以上的二级质谱序列分析方法可以推断ga+rn的结构。寡糖组分s3的碎片离子m/z485的质谱断裂模式示意图如图23右侧的视图b所示。
对寡糖组分s4的碎片离子m/z419的esi-cid-ms/ms谱如图24左侧的视图a所示。硫酸化葡萄糖醛酸鼠李二糖m/z419是由葡萄糖醛酸鼠李二糖硫酸酯化得到。在其二级质谱中,碎片离子m/z243的出现表明硫酸基团位于鼠李糖上,其结构推断为β-d-glca-(1→3)-α-l-rhap(2so4)。寡糖组分s4的碎片离子m/z419的质谱断裂模式示意图如图24右侧的视图b所示。
寡糖组分s4的碎片离子m/z565的esi-cid-ms/ms谱如图25左侧的视图a所示。硫酸化葡萄糖醛酸鼠李三糖m/z565由葡萄糖醛酸鼠李三糖硫酸化得到。其结构推断为β-d-glca-(1→3)-α-l-rhap(2so4)-(1→3)-α-l-rhap。寡糖组分s4的碎片离子m/z565的质谱断裂模式示意图如图25右侧的视图b所示。
以下为绿藻多糖的性能测试和分析,本发明的绿藻多糖能够显著延长活化部分凝血酶时间和凝血酶原时间,降低纤维蛋白原含量,说明其具有较好的抗凝血活性。本发明的绿藻多糖能够显著增加大鼠血浆纤维蛋白(原)降解产物和d-二聚体含量,降低纤溶酶原激活抑制物水平,增加血凝块溶解率,其活性高于尿激酶,说明其具有很好的纤溶和溶栓作用。另外,本发明的绿藻多糖能够增加人肝癌细胞株(以下将人肝癌细胞株简写为hepg2)细胞及胰岛素抵抗hepg2细胞的葡萄糖消耗能力,降低胰岛素抵抗hepg2细胞的甘油三酯和总胆固醇水平,其作用高于二甲双胍,表明其具有好的降血糖降血脂活性。综上所述,本发明的绿藻多糖具有明显抗凝血活性、纤溶和溶栓活性以及降血糖降血脂活性,可用于制备抗凝血和降血糖药物及保健食品。
本发明的绿藻多糖的抗凝血活性是通过测定体外活化部分凝血酶时间、凝血酶时间、凝血酶原时间和纤维蛋白原含量进行评价,以肝素作为阳性对照。活化部分凝血酶时间反映了内源性或者共同凝血途径,凝血酶时间反映了对凝血酶活性或者纤维蛋白原的作用,凝血酶原时间反映了外源性凝血途径,纤维蛋白原主要反映纤维蛋白原的含量。表1是绿藻多糖的抗凝血活性分析结果。由表1可知,本发明的绿藻多糖显著延长活化部分凝血酶时间和凝血酶原时间,降低纤维蛋白原含量,说明其具有较好的抗凝血活性,也说明绿藻多糖的抗凝血活性是通过抑制内源和共同凝血途径以及凝血酶活性起作用的,而对外源性凝血途径没有影响。
表1
本发明的绿藻多糖的纤溶和溶栓活性是通过体内纤维蛋白(原)降解产物、d-二聚体和纤溶酶原激活抑制物水平以及体外血凝块溶解率进行评价,以尿激酶作为阳性对照。d-二聚体以及纤维蛋白(原)降解产物水平是判断血凝以及纤溶系统情况的理想指标。在纤溶系统的平衡中,纤溶酶原激活剂及其抑制因子起着关键的调控作用,其中纤溶酶原激活抑制物是血浆中主要的纤溶酶原激活物抑制剂。由图26可知,绿藻多糖能使大鼠血浆的纤维蛋白(原)降解产物水平显著升高,在2.5毫克/千克时,已经能够极显著地增加纤维蛋白(原)降解产物的水平,且随着绿藻多糖剂量的增大,纤维蛋白(原)降解产物的含量逐渐升高,在10毫克/千克时,大鼠血浆的纤维蛋白(原)降解产物含量与阳性药尿激酶组存在极显著性差异。由图27可知,绿藻多糖随着其剂量的增加,能够显著提高大鼠血浆的d-二聚体的含量,在10毫克/千克时,大鼠血浆的d-二聚体含量与阳性药尿激酶组相比存在极显著性差异。由图28可知,绿藻多糖能够降低大鼠血浆的纤溶酶原激活抑制物水平,在2.5毫克/千克时,绿藻多糖就能显著降低大鼠血浆的纤溶酶原激活抑制物水平,在10毫克/千克时,纤溶酶原激活抑制物的水平降至0。由图29可知,随着绿藻多糖剂量的增加,大鼠血浆的血凝块溶解率逐渐增加,且在10毫克/毫升时,溶解率与阳性药尿激酶组相比具有极显著性差异,这些结果说明绿藻多糖具有很好的纤溶和溶栓活性。
本发明的绿藻多糖的降血糖降血脂活性是通过测定绿藻多糖对hepg2细胞及棕榈酸诱导的胰岛素抵抗hepg2细胞葡萄糖消耗能力、甘油三酯及总胆固醇水平的影响进行评价,以二甲双胍作为阳性对照。评价绿藻多糖的降血糖降血脂活性之前,首先测定绿藻多糖对hepg2细胞增殖的影响,确定了绿藻多糖的用药浓度范围。由图30可知,随着绿藻多糖浓度的增加,hepg2细胞的葡萄糖消耗能力逐渐增加,在50、100和200微克/毫升时,与空白组存在极显著性差异,且在200微克/毫升时,葡萄糖消耗能力达到最大,与二甲双胍组相似。由图31可知,模型组的葡萄糖消耗能力显著低于空白组,表明造模成功。绿藻多糖能够极显著地增加胰岛素抵抗hepg2细胞葡萄糖的消耗,且随着绿藻多糖浓度的增加,葡萄糖消耗逐渐增大,当浓度为200微克/毫升,与模型组相比,具有显著性差异。由图32可知,模型组的甘油三酯含量与空白组相比具有极显著性差异,表明造模成功。绿藻多糖能够极显著地降低胰岛素抵抗hepg2细胞中甘油三酯的含量,50微克/毫升时,降低作用达到最强,与二甲双胍的作用相似。由图33可知,与空白组相比,模型组的总胆固醇含量极显著地增加,表明造模成功。绿藻多糖能够极显著地降低胰岛素抵抗hepg2细胞中总胆固醇的含量,当绿藻多糖浓度为25微克/毫升和50微克/毫升时,降低总胆固醇的作用最强,优于二甲双胍的作用。
以上实施例仅用以说明本发明的技术方案,而非对其进行限制;尽管参照前述实施例对本发明进行了详细的说明,对于本领域的普通技术人员来说,依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或替换,并不使相应技术方案的本质脱离本发明所要求保护的技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!