4‑羟基环孢素的制备方法与流程
本发明涉及有机合成领域,特别是涉及一种4-羟基环孢素的制备方法。
背景技术:
4-羟基环孢素((γ-hydroxylmethylleucine4)-cyclosporina)最早是由山德士(sandoz)制药公司在1991年开发的一种aids抑制活性的环孢素免疫活性抑制衍生物。4-羟基环孢素是由环孢素的4位亮氨酸侧链γ位氧化获得的,结构式为
4-羟基环孢素为中间体在抗病毒分子的研发中得到广泛应用。如,scynexis公司研制的抗hcv临床药物分子scy-635正是在(γ-hydroxylmethylleucine4)-cyclosporina的基础上再经过3位修饰得到的。此外,(γ-hydroxylmethylleucine4)-cyclosporina本身还具有良好的生发作用。
目前已有的4-羟基环孢素的制备方法收率低,需要多步柱层析,纯化困难,很难进行产业化生产。因此急需找到一种可以大量制备4-羟基环孢素的制备方法,以便进行更深入的衍生化研究。
技术实现要素:
基于此,有必要提供一种4-羟基环孢素的制备方法,用于大量制备4-羟基环孢素。
一种4-羟基环孢素的制备方法,包括如下步骤:
步骤一、-20℃~35℃下将化合物2和第一反应物溶于第一有机溶剂中,接着在-15℃~30℃下加入3-氯-2-甲基丙烯,接着加热至58℃~82℃并充分反应,接着加入水并用酸性调节剂调节ph为7,充分反应后纯化得到化合物3,其中,所述第一反应物选自nah、kh、k2co和cs2co3中的至少一种,所述酸性调节剂为盐酸、柠檬酸、硫酸或氟硼酸,所述化合物2以及所述化合物3的结构式如下,
化合物2:
步骤二、将所述化合物3溶解于乙醇中,接着加入naoh溶液或koh溶液并在回流条件下充分反应,接着除去乙醇并用所述酸性调节剂调节ph至7,回流条件下充分反应,之后再用浓盐酸调节ph至3,纯化后得到化合物4,其中,所述化合物4的结构式如下,
化合物4:
步骤三、将所述化合物4溶解于ph为8的三羟甲基氨基甲烷盐酸盐的水溶液中,接着加热至30℃~55℃并加入酰化氨基酸水解酶,充分反应后加入吸附剂并过滤,保留滤液并加入等体积的thf、二氧六环或乙腈,接着降温至-5℃~10℃并加入二碳酸二叔丁酯,用碳酸氢钠调节ph为7.5~8.5后控制反应温度为-5℃~30℃并充分反应,然后除去thf,并用浓盐酸调节ph为3,接着纯化得到化合物5,其中,所述化合物5的结构式如下,
化合物5:
步骤四、将所述化合物5、碘甲烷和nah在干燥的第二有机溶剂中混合并充分反应,接着冷却至-10℃~5℃后纯化,接着加入水合肼并在室温下充分反应,再次纯化得到化合物6,其中,所述化合物6的结构式如下,
化合物6:
步骤五、-8℃~4℃下将所述化合物6溶解于第三有机溶剂中并降温至-15℃~-8℃,接着加入第二反应物或第三反应物,控制反应温度为-15℃~-8℃并充分反应,纯化后得到化合物7,其中,所述第二反应物为氯化氢的乙醚溶液和亚硝酸叔丁酯,所述第三反应物为乙酸和亚硝酸钠,所述化合物7的结构式如下,
化合物7:
步骤六、将环孢霉素a、吡啶和4-二甲氨基吡啶溶于乙酸酐中,室温下充分反应,接着纯化得到化合物8,其中,环孢霉素a和所述化合物8的结构式如下,
环孢霉素a:
化合物8:
步骤七、将所述化合物8和三甲基氧四氟硼酸盐溶于第四有机溶剂中并充分反应,接着加入乙腈和水并充分反应,纯化得到化合物9,其中,三甲基氧四氟硼酸盐和所述化合物9的结构式如下,
三甲基氧四氟硼酸盐:
化合物9:
步骤八、将所述化合物9、n,n-二异丙基乙胺和硫代异氰酸苯酯溶于第五有机溶剂中并充分反应,接着加入n,n-二甲基乙二胺并充分反应,接着加入甲醇和质量分数为40%~60%的氟硼酸水溶液并充分反应,之后纯化得到化合物10,其中,所述化合物10的结构式如下:
化合物10:
步骤九、在温度为-8℃~-12℃的条件下将所述化合物10、所述化合物7和n,n-二异丙基乙胺溶于第六有机溶剂中,接着升温至室温并充分反应,接着纯化得到化合物11,其中,所述化合物11的结构式如下,
化合物11:
步骤十、在温度为-20℃~8℃的条件下将所述化合物11和第四反应物溶解在四氢呋喃和水的混合溶液中,充分反应后纯化并在温度为-20℃~8℃的条件下加入二氯甲烷和三氟乙酸,充分反应后再次纯化,并在温度为-20℃~8℃的条件下加入n,n-二异丙基乙胺和bopcl,升温至室温并充分反应,第三次纯化后得到化合物12,其中,所述第四反应物为lioh、naoh或koh,bopcl和所述化合物12的结构式如下,
bopcl:
化合物12:
步骤十一、将所述化合物12、甲基磺酸和醋酸溶于第七有机溶剂中,控制温度为30℃~50℃并充分反应,纯化后得到化合物13,其中,所述化合物13的结构式如下,
化合物13:
步骤十二、将所述化合物13和甲醇钠溶于第八有机溶剂中并充分反应,纯化后得到化合物1,所述化合物1即为4-羟基环孢素,其中,所述化合物1的结构式如下,
化合物1:
在一个实施例中,步骤一中,所述第一有机溶剂为二甲基甲酰胺、氮甲基吡烙烷酮或thf。
在一个实施例中,步骤三中,所述纯化得到化合物5的操作为:萃取后保留有机相并洗涤,接着浓缩所述有机相并加入乙醚、甲基叔丁基醚或异丙醚,去除析出的化合物4’得到所述化合物5的乙醚溶液,其中,所述化合物4’的结构式如下,
化合物4’:
在一个实施例中,步骤四中,所述第二有机溶剂为四氢呋喃、二甲基甲酰胺或体积比为2:1的四氢呋喃和二甲基甲酰胺的混合溶液。
在一个实施例中,步骤五中,所述第三有机溶剂为四氢呋喃、二氧六环或dmf。
在一个实施例中,步骤七中,所述第四有机溶剂为二氯甲烷、二氯乙烷或氯仿。
在一个实施例中,步骤八中,所述第五有机溶剂为二氯甲烷、四氢呋喃或甲苯。
在一个实施例中,步骤九中,所述第六有机溶剂为氯仿、甲苯或二氯甲烷。
在一个实施例中,步骤十一中,所述第七有机溶剂为二氯甲烷、甲苯或二氯乙烷。
在一个实施例中,步骤十二中,所述第八有机溶剂为甲醇。
这种4-羟基环孢素的制备方法在制备4-羟基环孢素时,化学合成的速度快、成本低,操作简便,制得的4-羟基环孢素纯度较高,可以实现4-羟基环孢素的大量制备。
附图说明
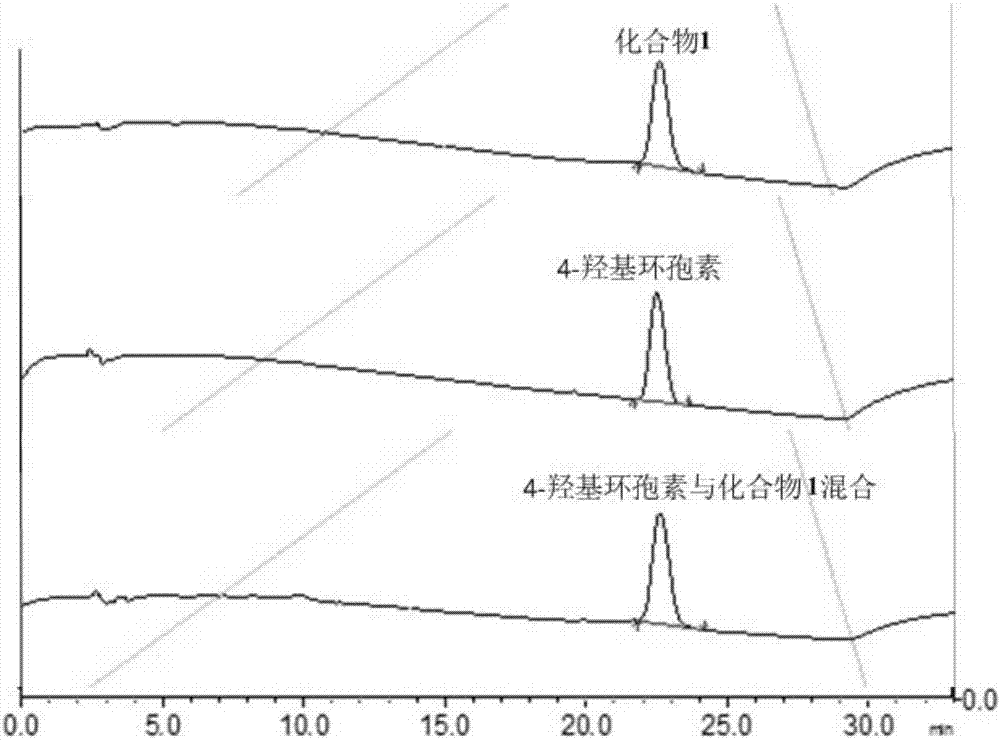
图1为实施例1制得的化合物1与标准4-羟基环孢素的比对测试图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
本发明公开了一实施方式的4-羟基环孢素的制备方法,其中,4-羟基环孢素((γ-hydroxylmethylleucine4)-cyclosporina)的结构式如下:
具体的,4-羟基环孢素的制备方法包括如下步骤:
步骤一、-20℃~35℃(优选为冰浴)下将化合物2和第一反应物溶于第一有机溶剂中,接着在-15℃~30℃(优选为冰浴)下加入3-氯-2-甲基丙烯,接着加热至58℃~62℃(优选为60℃)并充分反应(反应时间优选为2h),接着加入水并用酸性调节剂调节ph为7,充分反应后纯化得到化合物3。
第一反应物选自nah、kh、k2co和cs2co3中的至少一种.
酸性调节剂为盐酸、柠檬酸、硫酸或氟硼酸。优选的,酸性调节剂为盐酸(浓度为0.5mol/l~12mol/l)。
化合物2、3-氯-2-甲基丙烯以及化合物3的结构式如下,
化合物2:
步骤一中,nah和3-氯-2-甲基丙烯相对于化合物2过量加入,以保证化合物2完全反应。
优选的,化合物2、nah和3-氯-2-甲基丙烯的摩尔比为1:1.2:1.2。
第一有机溶剂可以为二甲基甲酰胺(dmf)、氮甲基吡烙烷酮或thf。
充分反应后纯化得到化合物3的操作可以为:充分反应后析出的白色固体经过过滤收集并用蒸馏水洗涤,最后真空干燥得到化合物3。
步骤二、将步骤一得到的化合物3溶解于乙醇中,接着加入naoh溶液(浓度为2mol/l~4mol/l,优选为3mol/l)或koh溶液(浓度为2mol/l~4mol/l,优选为3mol/l)并在回流条件下充分反应(反应时间优选为2h),接着除去乙醇并用上述酸性调节剂调节ph至7,回流条件下充分反应,之后再用浓盐酸(浓度优选为12mol/l)调节ph至3,纯化后得到化合物4。
化合物4的结构式如下,
化合物4:
步骤二中,其他反应物相对于化合物3过量加入,以保证化合物3完全反应。
纯化后得到化合物4的操作为:过滤后保留白色晶体,并在乙醚中重结晶得到化合物4。
步骤三、将步骤二得到的化合物4溶解于ph为8的三羟甲基氨基甲烷盐酸盐的水溶液(浓度为0.05m~1m)中,接着加热至30℃~55℃(优选为40℃)并加入酰化氨基酸水解酶,充分反应后加入吸附剂并过滤,保留滤液并加入等体积的thf、二氧六环或乙腈,接着降温至-5℃~10℃(优选为5℃)并加入二碳酸二叔丁酯,用碳酸氢钠调节ph为7.5~8.5后控制反应温度为-5℃~30℃(优选为10℃)并充分反应,然后除去thf,并用浓盐酸(浓度优选为12mol/l)调节ph为3,接着纯化得到化合物5。
化合物5的结构式如下,
化合物5:
步骤三中,其他反应物相对于化合物4过量加入,以保证化合物4完全反应。
吸附剂优选为硅藻土。
步骤三中,反应产物为化合物4’和化合物5,因此,为了得到化合物5,需要在纯化操作中除去化合物4’。
纯化得到化合物5的操作为:萃取(萃取剂优选为乙酸乙酯)后保留有机相并洗涤(优选为用10%的氯化钠溶液洗涤),接着浓缩所述有机相并加入乙醚、甲基叔丁基醚或异丙醚,去除析出的化合物4’得到化合物5的乙醚溶液。
化合物4’的结构式如下,
化合物4’:
步骤四、将步骤三得到的化合物5、碘甲烷和nah在干燥的第二有机溶剂中混合并充分反应(反应时间优选为12h),接着冷却至-10℃~5℃(优选为0℃)后纯化,接着加入水合肼并在室温下充分反应(反应时间优选为6h),再次纯化得到化合物6。
化合物6的结构式如下,
化合物6:
步骤四中,其他反应产物均相对于化合物5过量加入,以保证化合物5完全反应。
第二有机溶剂为体积比为2:1的四氢呋喃(thf)和二甲基甲酰胺(dmf)的混合溶液。
冷却至-10℃~5℃后纯化的操作为:冷却至-10℃~5℃后加入柠檬酸,接着萃取(萃取剂优选为乙酸乙酯)并保留有机相,干燥(干燥剂优选为硫酸钠)后浓缩并除去第二有机溶剂。
再次纯化得到化合物6的操作为:用水稀释,接着萃取(萃取剂优选为甲基叔丁基醚)并保留有机相,过滤(优选为采用硅胶过滤)、干燥后得到化合物6。
步骤五、-8℃~4℃(优选为冰浴)下将步骤四得到的化合物6溶解于第三有机溶剂中并降温至-15℃~-8℃(优选为-10℃),接着加入第二反应物或第三反应物,控制反应温度为-15℃~-8℃(优选为-10℃)并充分反应(反应时间优选为1h),纯化后得到化合物7。
第二反应物为浓度为0.1mol/l~0.5mol/l的氯化氢的乙醚溶液和亚硝酸叔丁酯(1~10当量,相对于化合物6),第三反应物为乙酸(2~30当量,相对于化合物6)和亚硝酸钠(1~10当量,相对于化合物6)。
化合物7的结构式如下,
化合物7:
步骤五中,其他反应产物均相对于化合物6过量加入,以保证化合物6完全反应。
第三有机溶剂可以为四氢呋喃、二氧六环或dmf。
纯化后得到化合物7的操作为:旋蒸除去第三有机溶剂,剩余物萃取(萃取剂优选为氯仿)后保留有机相,接着分别用冰水和质量百分数为5%的碳酸氢钠溶液洗涤,最后干燥得到化合物7。
步骤六、将环孢霉素a(csa)、吡啶和4-二甲氨基吡啶溶于乙酸酐中,室温下充分反应(反应时间优选为12h),接着纯化得到化合物8。
环孢霉素a和化合物8的结构式如下,
环孢霉素a:
化合物8:
步骤六中,其他反应物均相对于环孢霉素a(csa)过量,以保证环孢霉素a(csa)完全反应。
纯化得到化合物8的操作为:用1l乙酸乙酯稀释,接着用食盐水、饱和的氯化铵溶液和15%的碳酸氢钠溶液依次洗涤,有机相干燥(优选为用硫酸钠干燥)、浓缩,最后在2-甲基四氢呋喃和甲基叔丁基醚的混合溶液(5/1~1/5v/v)中重结晶得到的白色固体即为化合物8。
步骤七、将步骤六得到的化合物8和三甲基氧四氟硼酸盐溶于第四有机溶剂中并充分反应(反应时间优选为20h),接着加入乙腈和水并充分反应(反应时间优选为3h),纯化得到化合物9。
三甲基氧四氟硼酸盐和化合物9的结构式如下,
三甲基氧四氟硼酸盐:
化合物9:
步骤七中,其他反应物均相对于化合物8过量,以保证化合物8完全反应。
第四有机溶剂可以为二氯甲烷、二氯乙烷或氯仿。
纯化得到化合物9的操作为:待分层后保留有机相,有机相用水洗涤(优选为洗涤3次),接着真空浓缩、干燥,最后在体积比为2:1的2-甲基四氢呋喃和甲基叔丁基醚的混合溶液中结晶,得到的白色晶状粉末为化合物9。
步骤八、将步骤七得到的化合物9、n,n-二异丙基乙胺和硫代异氰酸苯酯溶于第五有机溶剂中并充分反应(反应时间优选为100min),接着加入n,n-二甲基乙二胺并充分反应(反应时间优选为1h),接着加入甲醇和质量分数为40%~60%的氟硼酸水溶液(氟硼酸水溶液的质量分数优选为50%)并充分反应(反应时间优选为1h),之后纯化得到化合物10。
化合物10的结构式如下:
化合物10:
步骤八中,其他反应物均相对于化合物9过量,以保证化合物9完全反应。
第五有机溶剂为二氯甲烷、四氢呋喃或甲苯。
纯化得到化合物10的操作为:加水待分层后保留无机层,无机层用萃取剂(萃取剂优选为二氯甲烷)萃取,保留有机相并浓缩后,最后在体积比为2:1的2-甲基四氢呋喃和甲基叔丁基醚的混合溶液中重结晶,得到的白色泡沫状物质为化合物10。
步骤九、在温度为-8℃~-12℃(优选为-10℃)的条件下将步骤八得到的化合物10、步骤五得到的化合物7和n,n-二异丙基乙胺溶于第六有机溶剂中,接着升温至室温并充分反应(反应时间优选为3h),接着纯化得到化合物11。
化合物11的结构式如下:
化合物11:
步骤九中,其他反应物均相对于化合物10过量,以保证化合物10完全反应。
第六有机溶剂可以为氯仿、甲苯或二氯甲烷。
纯化得到化合物11的操作为:旋蒸除去第六有机溶剂,剩余物溶解在乙酸乙酯中,用稀盐酸(浓度为0.5mol/l~2mol/l,优选为1mol/l)和水洗涤有机相,干燥并浓缩后得到化合物11。
步骤十、在温度为-20℃~8℃的条件下将步骤九得到的化合物11和第四反应物溶解在体积比为10:1~1:3的四氢呋喃和水的混合溶液中,充分反应(反应时间优选为2h)后纯化,并在温度为-20℃~8℃(优选为0℃)的条件下加入体积比为20~5:1(优选为10:1)的二氯甲烷和三氟乙酸,充分反应(反应时间优选为4h)后再次纯化,并在温度为-20℃~8℃的条件下加入n,n-二异丙基乙胺和bopcl,升温至室温并充分反应,第三次纯化后得到化合物12。
第四反应物为lioh、naoh或koh。
bopcl和所述化合物12的结构式如下:
bopcl:
化合物12:
步骤十中,其他反应物均相对于化合物11过量,以保证化合物11完全反应。
充分反应后纯化的操作为:充分反应后用二氯甲烷稀释并保留有机相,接着分别用1n的盐酸、水和食盐水洗涤有机相,最后浓缩有机相。
充分反应后再次纯化的操作为:充分反应后用二氯甲烷稀释并保留有机相,接着分别用碳酸氢钠饱和溶液以及饱和食盐水洗涤有机相,有机相干燥后浓缩。
第三次纯化后得到化合物12的操作为:用二氯甲烷稀释并保留有机相,接着依次用水、1n的氯化氢溶液、水和饱和食盐水洗涤,有机相干燥浓缩后在体积比为2:1的2-甲基四氢呋喃和甲基叔丁基醚的混合溶液结晶,得到的白色泡沫状物质为化合物12。
步骤十一、将步骤十得到的化合物12、甲基磺酸和醋酸溶于第七有机溶剂中,控制温度为30℃~50℃(优选为40℃)并充分反应(反应时间优选为8h),纯化后得到化合物13。
化合物13的结构式如下:
化合物13:
步骤十中,其他反应物均相对于化合物12过量,以保证化合物12完全反应。
第七有机溶剂可以为二氯甲烷、甲苯或二氯乙烷。
纯化后得到化合物13的操作为:用二氯甲烷稀释后保留有机相,接着依次用水、碳酸氢钠溶液、水以及饱和食盐水洗涤有机相,有机相经干燥、浓缩后得到化合物13。
步骤十二、将步骤十一得到的化合物13和甲醇钠溶于第八有机溶剂中并充分反应(反应时间优选为15h),纯化后得到化合物1,化合物1即为4-羟基环孢素。
化合物1(4-羟基环孢素)结构式如下:
化合物1:
步骤十中,其他反应物均相对于化合物13过量,以保证化合物13完全反应。
第八有机溶剂为甲醇。
纯化后得到化合物1的操作为:用0.5n的盐酸将ph调至6左右,浓缩反应液后得到的浓缩物溶解在乙酸乙酯中,接着依次用碳酸氢钠和食盐水洗涤有机相,有机相浓缩后经色谱柱分离纯化,得到化合物1。
这种4-羟基环孢素的制备方法在制备4-羟基环孢素时,化学合成的速度快、成本低,操作简便,制得的4-羟基环孢素纯度较高,可以实现4-羟基环孢素的大量制备。
以下为具体实施例。
具体实施例中用到的药品和仪器均为本领域常规选择。
实施例1
步骤一、将化合物2(乙酰氨基丙二酸二乙酯,217.1g,1.0mol)溶于2.0ldmf中,随后加入nah(60wt%溶在煤油中,48.0g,1.2mol)。然后在冰浴下加入3-氯-2-甲基丙烯(108.7g,117.5ml,1.2mol),在30min内加完。之后将温度加热到60摄氏度,并保持2小时。随后加入水(2.0l),再用1.0mol/lhcl将溶液的ph值调至7。在室温下,反应12h,析出的白色固体经过过滤收集并用蒸馏水洗涤(2×1l),最后用真空干燥箱干燥,得到化合物3(247.0g,91%)。
步骤二、将化合物3(108.6g,0.5mol)溶于0.5l乙醇中,加入0.5l的3.0mnaoh的水溶液,反应回流3h,然后除去乙醇,再用浓盐酸(12mol/l)调至ph值为7,回流2h,之后再用浓盐酸(12mol/l)将溶液的ph值调至3,析出的白色固体经过过滤收集,并在乙醚中重结晶得化合物4(71.0g,83%)。
步骤三、将化合物4(17.1g,100.0mmol)溶于ph8.0、浓度为0.1m的三羟甲基氨基甲烷盐酸盐的水溶液中,将温度调至40℃,加入酰化氨基酸水解酶(l-aminoacylase,0.86g,3units/gsubstrate),反应5h。加入26g硅藻土,过滤。向滤液中加入等体积的thf,并将温度冷却至5℃。随后加入二碳酸二叔丁酯(boc2o,13.1g,60.0mol),用碳酸氢钠将ph控制为8。反应温度控制在10℃下搅拌反应4h,然后除去thf。接着用浓盐酸(12mol/l)将水相酸化至ph为3,用乙酸乙酯萃取3次,每次300ml。将乙酸乙酯层的有机相合并,并用10%的氯化钠溶液洗涤,浓缩并加入乙醚,去除析出的化合物4’得到化合物5的乙醚溶液,去除乙醚得到亮黄色的油状产物化合物5(10.8g,47%,>99%ee)。
步骤四、将化合物5(10.8g,4.7mmol)和碘甲烷(1.8ml,28.5mmol)溶于干燥的thf-dmf(2:1,15ml)中,加入氢化钠(60wt%,564mg,14.1mmol)。反应过夜,再将冷至0℃,加入柠檬酸(12ml),并用乙酸乙酯萃取三次,每次100ml。合并有机相,用硫酸钠干燥,过滤,浓缩除去四氢呋喃和乙酸乙酯。再加入水合肼(90%,2.6ml,溶于10mldmf)。再将反应在室温下搅拌6小时。用100ml水稀释和甲基叔丁基醚萃取(2×100ml)。合并有机相,经硅胶过滤,干燥,得到化合物6(8.9g,74%)。
步骤五、在冰浴中,将化合物6(0.57g,2.2mmol)溶解在四氢呋喃(5ml)中,温度降到-10℃。加入氯化氢的乙醚溶液(14.0m,0.8ml)和亚硝酸叔丁酯(0.38ml,3.0mmol)。将反应液在零下10℃中搅拌60分钟,直至溶液变澄清。然后,经旋蒸除去溶剂,剩余物用氯仿萃取,接着用冰水和5%的碳酸氢钠溶液,冰水洗涤,最后干燥得到化合物7。
步骤六、将环孢霉素a(csa,24.0g,20.0mmol)溶于80ml乙酸酐中,加入80ml吡啶和4-二甲氨基吡啶(dmpa,4-n,n-dimethylaminopyride,0.8g,6.5mmol)。将反应置于室温并搅拌过夜,接着用1l乙酸乙酯稀释。再用食盐水,饱和的氯化铵溶液和质量分数为15%的碳酸氢钠溶液依次洗涤。有机相用硫酸钠干燥,过滤,在真空下浓缩。在2-甲基四氢呋喃(100ml)和甲基叔丁基醚(50ml)的混合溶液中重结晶得到的白色固体为化合物8(乙酰基-环孢霉素a,23.2g,93%)。
步骤七、将化合物8(37.4g,30mmol)溶于二氯甲烷(300ml),加入三甲基氧四氟硼酸盐(13.3g,90mmol)。反应20小时后加入乙腈和水,再反应3小时,分层,有机相用水洗涤3次,每次用100ml。接着,将反应液经真空浓缩、干燥。然后在2-甲基四氢呋喃(100ml)和甲基叔丁基醚(50ml)的混合溶液中结晶得到的白色晶状粉末为化合物9(29.9g,73%)。
步骤八、将化合物9(13.7g,10.0mmol)溶于二氯甲烷(100ml)中,加入n,n-二异丙基乙胺(6.6ml,40mmol)和硫代异氰酸苯酯(4.1g,30mmol),反应搅拌100分钟,加入n,n-二甲基乙二胺(2.3ml,25mmol),接着搅拌60分钟,然后加入甲醇(60ml)和50%的氟硼酸水溶液(2.3g,13mmol)。反应搅拌1小时。然后,加入水(50ml),分层,无机层用二氯甲烷萃取2次。合并有机相,浓缩后,在2-甲基四氢呋喃(50ml)和甲基叔丁基醚(25ml)的混合溶液中重结晶,得到的白色泡沫状物质为化合物10(11.2g,91%)。
步骤九、将化合物10(2.47g,2.0mmol)溶解在氯仿(10ml)中。随后在零下10℃加入化合物7和n,n-二异丙基乙胺,再将温度恢复到室温,搅拌3小时。经旋蒸除去溶剂,剩余物溶解在乙酸乙酯中,用稀盐酸(1mol/l)和水洗涤有机相,干燥并浓缩后得到化合物11(2.6g,95%)。
步骤十、将化合物11(2.6g,1.9mmol)溶解在体积比为2:1的四氢呋喃和水的混合溶液中,在0℃中,加入氢氧化锂(385mg,9.5mmol),反应搅拌2小时。然后用二氯甲烷稀释,分别用1.0n的盐酸、水和食盐水洗涤。有机相浓缩后在0℃下加入20ml二氯甲烷和5ml的三氟乙酸。反应搅拌4小时,用二氯甲烷稀释后,用碳酸氢钠饱和以及饱和食盐水洗涤有机相。有机相干燥后浓缩后在0℃下加入n,n-二异丙基乙胺(4.8ml,9.5mmol)和bopcl(967mg,3.8mmol)。将温度调至室温,反应搅拌5小时,用二氯甲烷稀释,依次用水、1.0n的氯化氢溶液、水和饱和食盐水洗涤。有机相干燥浓缩后在2-甲基四氢呋喃(6.67ml)和甲基叔丁基醚(3.33ml)的混合溶液结晶,得到的白色泡沫状物质为化合物12(1.18g,62%)。
步骤十一、将化合物12(248mg,0.2mmol)溶解在二氯甲烷(2ml)中,加入甲基磺酸(msa,13ul,0.2mmol)和醋酸(114ul,2.0mmol)。反应在40℃搅拌8小时,用二氯甲烷稀释,依次用水、碳酸氢钠溶液、水以及饱和食盐水洗涤有机相。有机相经干燥、浓缩后得到化合物13(244mg,95%)。
步骤十二、将化合物13(128mg,0.1mmol)溶于甲醇(5ml)中,加入甲醇钠的甲醇溶液(0.5m,0.5ml)。搅拌15小时后用0.5n的盐酸将ph调至6左右,反应液浓缩后,浓缩物溶解在50ml的乙酸乙酯中,用碳酸氢钠和食盐水洗涤。有机相浓缩后经色谱柱分离纯化,得到目的产物(97mg,80%)。
对得到的化合物1与标准4-羟基环孢素比对测试,结果如图1所示。
由图1可以看出,实施例1制得的化合物1即为4-羟基环孢素(γ-hydroxylmethylleucine4)-cyclosporina)。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!