一种炎症抑制化合物及其制备方法与流程
本发明涉及一种含氮稠环化合物及其制备方法,更特别地涉及一种炎症抑制化合物及其制备方法,属于有机化学合成领域。
背景技术
含氮杂环化合物一般都具有一定的生物活性和独特性质,从而在医药、农药、有机发光等领域中有着广泛的应用和研究前景。
作为含氮杂环化合物的一种,喹唑啉类化合物广泛存在于多种天然产物中,并通常具有良好的生物活性,例如除草、杀菌、杀虫、抗病毒、杀虫、抗病毒、抗炎、抗高血压、抗痉挛、抗肿瘤、抗疟疾和抗结核,此外还可应用到有机发光技术领域,从而在医药、农业、冶金等都多个领域都有着良好的应用前景和潜力。
迄今为止,科学家已发现了该类衍生物在治疗领域中的多种特定靶点应用,对于多种疾病致病因子具有优异的抑制作用。例如现有技术中已发现2-三氯甲基-4-芳硫基喹唑啉衍生物具有良好的抗疟疾活性(见bioorg.med.chem.lett.,21,p6003-6006,2011年),而有些4-杂芳硫基喹唑啉衍生物对某些癌细胞具有抗增殖活性(见bioorg.med.chem.lett.,17,p2193-2196,2007年),某些喹唑啉类化合物对表皮生长因子受体(egfr)和酪氨酸激酶(egfr-tk)有着很强的抑制作用,可用于抗癌。更有多种喹唑啉化合物作为药物已经上市销售,例如降血压药哌唑嗪、利尿药甲酸喹唑酮、抗肠癌药雷替曲塞、抗疟疾药常山碱、抗癌药物吉非替尼、杀菌药物丙氧喹啉等。
正是由于喹唑啉类化合物的广泛的应用前景和潜在治疗作用,其相应化合物的合成和新型喹唑啉类化合物的的寻求与研究业已成为有机化学合成的一个研究热点和重点。
例如,2-芳基喹唑啉类化合物的制备方法按照反应条件的不同而可分为两类:第一类是金属参与的催化成环反应,主要是采用过渡金属钯、铜等作为催化剂,使有机胺与相应的芳醛或者酰胺化合物环合而成。第二类则是使用强氧化剂如ddq、mno2、naclo等去氧化成环。
又如:
cn103242299a公开了如下新型喹唑啉衍生物、制备方法及其在有机电致发光中的用途:
上述两个化合物分别是由2-(4-溴苯)-4-苯基喹唑啉通过ullman反应与咔唑和二苯胺反应而得到。
rupamsarma等人(“microwave-promotedefficientsynthesisiofdihydroquinazolines”,greenchemistry,2011,13,718-722)报道了在微波辅助下,邻氨基芳酮、醛和尿素一起反应,从而得到喹唑啉化合物的方法,其反应式如下:
2010年,fu等人以乌尔曼偶联反应模型,使用更经济的酰胺为反应底物合成了2-取代喹唑啉化合物,反应式如下:
danzhao等人(“potassiumiodide-catalyzedthree-componentsynthesisof2-arylauinazolinesviaaminationofbenzylicofc-hbondsofmethylarenes”,advancedsynthesis&catalysis,2015,357,339-344)中公开了一种由邻氨基芳醛制备喹唑啉化合物的方法,其反应式如下:
cn102321075b公开了由式(ii)化合物与式(iii)反应,然后再与咪唑在固体碳酸钾催化下反应而制备如下通式(i)喹唑啉衍生物的方法:
cn103113311a公开了2-芳基喹唑啉或2-杂芳基喹唑啉化合物的制备方法,所述方法首先使芳基醛或杂芳基醛与邻氨基苯甲酰胺反应,得到2-芳基喹唑啉酮或2-杂芳基喹唑啉酮,然后经过还原得到2-芳基喹唑啉或2-杂芳基喹唑啉,其反应式如下:
wuzhang等人(“synthesisofquinazolinesviacuonanoparticlescatalyzedaerobicoxidativecouplingofaromaticalcoholsandamidines”,organic&biomolecularchemistry,2014,12,5752-5756)报道了氧化铜催化的喹唑啉化合物的合成方法,其反应式如下:
wang等人使用i2/tbhp作为氧化体系,以2-氨基二苯甲酮和苄胺为原料而合成了二取代喹唑啉化合物,其反应式如下:
如上所述,现有技术中公开了多种新型的喹唑啉化合物及其合成方法,但为了得到更多的、具有药物活性的喹唑啉化合物并对其合成方法进行研究,仍仍存在继续进行研究和探索的必要,这也是有机合成技术领域中的研究热点和重点之一。
技术实现要素:
为了寻求新型的具有药物活性的喹唑啉化合物及其制备方法,本发明人对此进行了深入研究,在付出了大量创造性劳动后,从而完成了本发明。
具体而言,第一个方面,本发明的技术方案和内容涉及一种下式(7)所示炎症抑制化合物:
其中,r1-r3各自独立地选自h、卤素、c1-c6烷基、卤代c1-c6烷基、c1-c6烷氧基或卤代c1-c6烷氧基。
在本发明的所述化合物中,所述卤素为卤族元素,例如可为f、cl、br或i。
在本发明的所述化合物中,所述c1-c6烷基的含义是指具有1-6个碳原子的直链或支链烷基,非限定性地例如可为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基或正己基等。
在本发明的所述化合物中,所述c1-c6烷氧基的含义是指具有上述含义的c1-c6烷基与氧原子相连后得到的基团。
在本发明的所述化合物中,所述卤代c1-c6烷基的含义是指具有上述含义的卤素与上述含义的c1-c6烷基相连后得到的基团。
在本发明的所述化合物中,所述卤代c1-c6烷氧基的含义是指具有上述含义的卤素与上述含义额c1-c6烷氧基相连后得到的基团。
所述式(7)炎症抑制化合物是一种全新的化合物,因具有喹唑啉环以及活性基团氨基,从而如上述“背景技术”中所述,可以后续合成更多的喹唑啉类化合物,在多个技术领域具有良好的应用开发潜力和研究价值。以及经过本发明人的研究发现,该化合物和中间体化合物具有明显的炎症抑制效果,在药物领域具有良好的研究前景和应用潜力。
第二个方面,本发明的技术方案和内容涉及所述式(7)炎症抑制化合物的制备方法,所述制备方法的路线如下:
其中,r1-r3的定义如上,在此不再一一赘述;x为卤素,该“卤素”的含义是指卤族元素,非限定地例如可为f、cl、br或i。
所述制备方法包括如下步骤:
s1:上式(1)化合物和上式(2)化合物在有机溶剂中,于碳酸钾存在下进行反应,反应结束后经后处理,得到上式(3)化合物;
s2:在有机溶剂中,于钯催化剂、有机配体和酸性添加剂作用下,上式(3)化合物和上式(4)化合物进行反应,反应结束后经后处理而得到所述式(5)化合物;
s3:上式(5)和上式(6)化合物在有机溶剂中,于碳酸钾存在下进行反应,反应结束后经后处理,得到上式(7)化合物。
下面,对各个步骤中的各个技术特征进行详细的进一步描述,具体如下。
[步骤s1]
在步骤s1中,所述有机溶剂为二甲基亚砜(dmso)。
其中,所述有机溶剂的用量并没有严格的限定,本领域技术人员可根据实际情况进行合适的选择与确定,例如其用量大小以方便反应进行和后处理即可,在此不再进行详细描述。
在步骤s1中,所述式(1)化合物与所述式(2)化合物的摩尔比为1:0.5-1.5,例如可为1:0.5、1:1或1:1.5。
在步骤s1中,所述式(1)化合物与碳酸钾的摩尔比为1:2-3,例如可为1:2、1:2.5或1:3。
在步骤s1中,反应温度为80-120℃,例如可为80℃、90℃、100℃、110℃或120℃。
在步骤s1中,反应时间为8-16小时,例如可为8小时、10小时、12小时、14小时或16小时。
在步骤s1中,反应结束后的所述后处理具体如下:反应结束后,将反应混合物倾入水中,并用乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比50:1的二氯甲烷和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到上式(3)化合物。
其中,在所述硅胶快速柱色谱提纯过程中,可通过tlc跟踪监控而确定合适的洗脱终点。
[步骤s2]
在步骤s2中,所述钯催化剂为乙酸钯(pd(oac)2)、乙酰丙酮钯(pd(acac)2)、氯化钯、三(二亚苄基丙酮)二钯(pd2(dba)3)或三氟乙酸钯(pd(tfa)2)中的任意一种,最优选为乙酸钯(pd(oac)2)。
在步骤s2中,所述有机配体为下式l1-l6中的任意一种,
所述有机配体最优选为l1。
在步骤s2中,所述酸性添加剂为对甲苯磺酸一水合物、乙酸、三氟乙酸、苯甲酸、甲烷磺酸、三氟甲烷磺酸或樟脑磺酸中的任意一种,优选为对甲苯磺酸一水合物或樟脑磺酸,最优选为对甲苯磺酸一水合物。
在步骤s2中,所述有机溶剂为四氢呋喃(thf)、n,n-二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、乙醇、1,4-二氧六环、二氯甲烷、环己烷或甲苯中的任意一种,优选为甲苯、二氯甲烷或环己烷,最优选为甲苯。
其中,所述有机溶剂的用量并没有严格的限定,本领域技术人员可根据实际情况进行合适的选择与确定,例如其用量大小以方便反应进行和后处理即可,在此不再进行详细描述。
在步骤s2中,所述式(3)化合物与所述式(4)化合物的摩尔比为1:1.5-2.5,例如可为1:1.5、1:2或1:2.5。
在步骤s2中,所述式(3)化合物与所述钯催化剂的摩尔比为1:0.02-0.1,例如可为1:0.02、1:0.04、1:0.06、1:0.08或1:0.1。
在步骤s2中,所述式(3)化合物与所述有机配体的摩尔比为1:0.05-0.15,例如可为1:0.05、1:0.1或1:0.15。
在步骤s2中,所述式(3)化合物与所述酸性添加剂的摩尔比为1:8-12,例如可为1:8、1:10或1:12。
在步骤s2中,反应温度为70-90℃,非限定性地例如可为70℃、80℃或90℃。
在步骤s2中,反应时间并无特别的限定,例如可通过液相色谱或tlc检测原料的残留量多少而确定合适的反应时间,例如可为16-28小时,非限定性地例如为16小时、20小时、24小时或28小时。
在步骤s2中,反应结束后的所述后处理可具体如下:反应结束后,将反应体系自然冷却至室温,用饱和碳酸钾水溶液洗涤,再加入乙酸乙酯萃取三次,合并有机相,用无水na2so4干燥,减压蒸馏,残留物通过硅胶快速柱色谱(以体积比16:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到上式(5)化合物。
其中,在所述硅胶快速柱色谱提纯过程中,可通过tlc跟踪监控而确定合适的洗脱终点。
[步骤s3]
在步骤s3中,所述有机溶剂为二氯甲烷。
其中,所述有机溶剂的用量并没有严格的限定,本领域技术人员可根据实际情况进行合适的选择与确定,例如其用量大小以方便反应进行和后处理即可,在此不再进行详细描述。
在步骤s3中,所述式(5)化合物与所述式(6)化合物的摩尔比为1:0.8-1.2,例如可为1:0.8、1:1或1:1.2。
在步骤s3中,所述式(5)化合物与碳酸钾的摩尔比为1:0.8-1.2,例如可为1:0.8、1:1或1:1.2。
在步骤s3中,反应温度为室温。
在步骤s3中,反应时间为3-7小时,例如可为3小时、4小时、5小时、6小时或7小时。
在步骤s3中,反应结束后的所述后处理具体如下:反应结束后,将反应混合物用水洗涤,并用足量乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比8:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到上式(7)化合物。
其中,在所述硅胶快速柱色谱提纯过程中,可通过tlc跟踪监控而确定合适的洗脱终点。
如上所述,本发明提供了一种所述式(7)炎症抑制化合物及其制备方法,所述化合物是一种全新的化合物,其具有喹唑啉环以及活性基团氨基等,可以后续合成更多的喹唑啉类化合物,而且该化合物及其中间体具有明显的炎症抑制效果,从而多个技术领域具有良好的应用开发潜力和研究价值。
附图说明
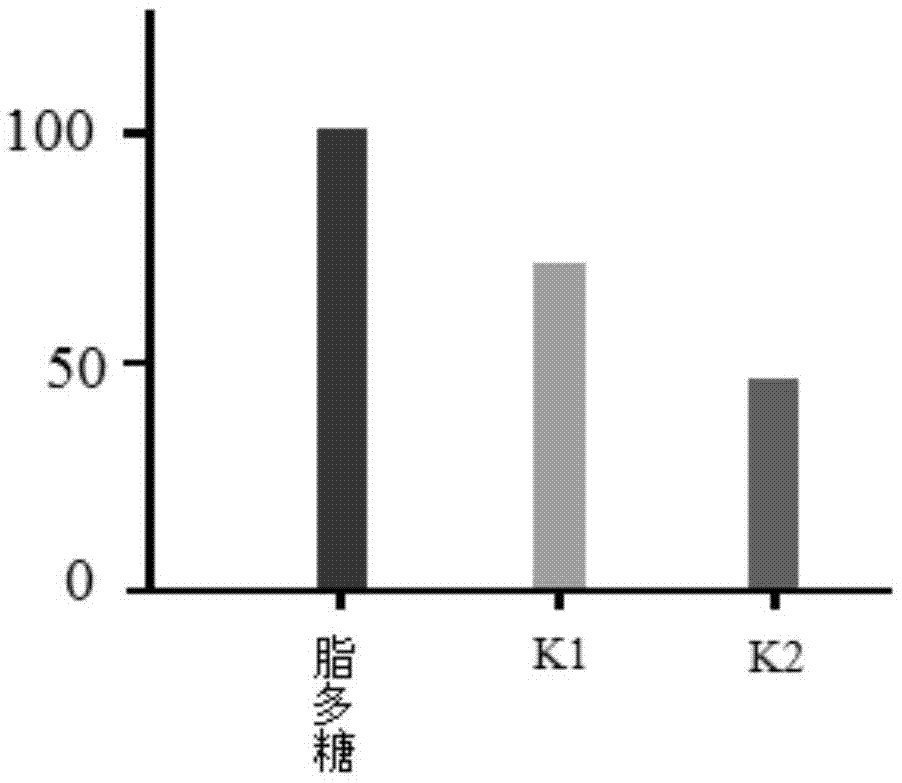
附图1是本发明化合物k1-k2对il-6的抑制效果图。
具体实施方式
下面通过具体实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,更非将本发明的保护范围局限于此。
实施例1
反应路线为:
具体包括如下步骤:
s1:向反应容器中的适量有机溶剂二甲基亚砜(dmso)中,加入100mmol上式(1)化合物、50mmol上式(2)化合物和300mmol碳酸钾,在80℃下搅拌反应16小时;
反应结束后,将反应混合物倾入水中,并用乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比50:1的二氯甲烷和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为白色固体的上式(3)化合物,产率为96.1%。
1hnmr(500mhz,dmso-d6)δ8.46(s,1h),8.24(d,j=8hz,1h),8.12(d,j=7.5hz,1h),7.93-7.98(m,2h),7.86(d,j=8hz,1h),7.75-7.82(m,2h),7.66(t,j=7.5hz,1h)。
s2:室温下,向适量有机溶剂甲苯中加入100mmol上式(3)化合物、150mmol上式(4)化合物、10mmol乙酸钯、5mmol有机配体l1和1200mmol对甲苯磺酸一水合物,然后搅拌升温至70℃并在该温度下搅拌反应28小时;
反应结束后,将反应体系自然冷却至室温,用饱和碳酸钾水溶液洗涤,再加入乙酸乙酯萃取三次,合并有机相,用无水na2so4干燥,减压蒸馏,残留物通过硅胶快速柱色谱(以体积比16:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为黄色固体的上式(5)化合物,将其命名为k1,产率为89.5%。
熔点:147-148℃。
1hnmr(500mhz,dmso-d6)δ8.56(d,j=8.5hz,1h),8.11(d,j=8.5hz,1h),8.02(d,j=8.5hz,1h),7.97(t,j=7.5hz,1h),7.84-7.86(m,2h),7.61-7.65(m,4h),7.49(s,2h),7.20(t,j=8.5hz,1h),6.87(d,j=8.5hz,1h),6.64(t,j=7.5hz,1h)。
s3:向反应容器中的适量有机溶剂二氯甲烷中,加入100mmol上式(5)化合物、80mmol上式(6)化合物和120mmol碳酸钾,在室温下搅拌反应3小时;
反应结束后,将反应混合物用水洗涤,并用足量乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比8:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的上式(7)化合物,将其命名为k2,产率为98.6%。
熔点:188-189℃。
1hnmr(500mhz,dmso-d6)δ13.52(s,1h),8.77-8.81(m,2h),8.09-8.15(m,3h),7.97(d,j=7.5hz,2h),7.89(d,j=7hz,2h),7.79(t,j=7.5hz,1h),7.60-7.71(m,5h),7.49(t,j=8hz,2h),7.33(t,j=7.5hz,1h)。
实施例2
反应路线同实施例1。
具体包括如下步骤:
s1:向反应容器中的适量有机溶剂二甲基亚砜(dmso)中,加入100mmol所述式(1)化合物、150mmol所述式(2)化合物和200mmol碳酸钾,在120℃下搅拌反应8小时;
反应结束后,将反应混合物倾入水中,并用乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比50:1的二氯甲烷和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为白色固体的所述式(3)化合物,产率为95.8%。
核磁表征数据同实施例1步骤s1的式(3)化合物。
s2:室温下,向适量有机溶剂甲苯中加入100mmol所述式(3)化合物、250mmol所述式(4)化合物、2mmol乙酸钯、15mmol有机配体l1和800mmol对甲苯磺酸一水合物,然后搅拌升温至90℃并在该温度下搅拌反应16小时;
反应结束后,将反应体系自然冷却至室温,用饱和碳酸钾水溶液洗涤,再加入乙酸乙酯萃取三次,合并有机相,用无水na2so4干燥,减压蒸馏,残留物通过硅胶快速柱色谱(以体积比16:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为黄色固体的所述式(5)化合物,产率为88.9%。
熔点与核磁表征数据同实施例1步骤s2的式(5)化合物。
s3:向反应容器中的适量有机溶剂二氯甲烷中,加入100mmol所述式(5)化合物、120mmol所述式(6)化合物和80mmol碳酸钾,在室温下搅拌反应7小时;
反应结束后,将反应混合物用水洗涤,并用足量乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比8:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的所述式(7)化合物,产率为98.8%。
熔点与核磁表征数据同实施例1步骤s3的式(7)化合物。
实施例3
反应路线同实施例1。
具体包括如下步骤:
s1:向反应容器中的适量有机溶剂二甲基亚砜(dmso)中,加入100mmol所述式(1)化合物、100mmol所述式(2)化合物和250mmol碳酸钾,在100℃下搅拌反应12小时;
反应结束后,将反应混合物倾入水中,并用乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比50:1的二氯甲烷和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为白色固体的所述式(3)化合物,产率为96.4%。
核磁表征数据同实施例1步骤s1的式(3)化合物。
s2:室温下,向适量有机溶剂甲苯中加入100mmol所述式(3)化合物、200mmol所述式(4)化合物、6mmol乙酸钯、10mmol有机配体l1和1000mmol对甲苯磺酸一水合物,然后搅拌升温至80℃并在该温度下搅拌反应22小时;
反应结束后,将反应体系自然冷却至室温,用饱和碳酸钾水溶液洗涤,再加入乙酸乙酯萃取三次,合并有机相,用无水na2so4干燥,减压蒸馏,残留物通过硅胶快速柱色谱(以体积比16:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为黄色固体的所述式(5)化合物,产率为89.4%。
熔点与核磁表征数据同实施例1步骤s2的式(5)化合物。
s3:向反应容器中的适量有机溶剂二氯甲烷中,加入100mmol所述式(5)化合物、100mmol所述式(6)化合物和100mmol碳酸钾,在室温下搅拌反应5小时;
反应结束后,将反应混合物用水洗涤,并用足量乙酸乙酯萃取两次,合并有机相,用水洗涤并用无水na2so4干燥,减压蒸馏,将所得残留物通过硅胶快速柱色谱(以体积比8:1的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的所述式(7)化合物,产率为98.9%。
熔点与核磁表征数据同实施例1步骤s3的式(7)化合物。
下面对步骤s2中的一些技术特征进行考察,从而对最优选条件进行了创造性选择,具体如下。
步骤s2中技术特征的考察
催化剂的考察
对比例s201-s203:除分别将步骤s2中的催化剂乙酸钯替换为乙酰丙酮钯(pd(acac)2)外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s201-s203。
对比例s204-s206:除分别将步骤s2中的催化剂乙酸钯替换为氯化钯外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s204-s206。
对比例s207-s209:除分别将步骤s2中的催化剂乙酸钯替换为三(二亚苄基丙酮)二钯(pd2(dba)3)外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s207-s209。
对比例s210-s212:除分别将步骤s2中的催化剂乙酸钯替换为三氟乙酸钯(pd(tfa)2)外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s210-s212。
结果见下表1(其中产物产率是指步骤s2中化合物(5)的产率)。
表1
其中,各个对比例与产物产率具有相应的对应关系,例如就对比例s201-s203而言,s201的产物产率为52.4%、s202的产物产率为52.9%和s203的产物产率为52.3%,其余以及下面表格中的如此表述也均有类似的对应关系,在此不再一一赘述。
由此可见,对于步骤s2中的催化剂而言,稍微的改变都可导致效果上的显著改变,例如当为三氟乙酸钯时,虽然与乙酸钯高度类似,但其产率仍有显著降低。这证明催化剂在种类上的选择具有不可预测性和效果上的非显而易见性。
有机配体的考察
除分别将有机配体l1替换为下表2中的其它有机配体外,其它操作均不变,从而重复实施了实施例1-3,所使用的有机配体、实施例对应关系和产物产率见下表2(其中产物产率是指步骤s2中化合物(5)的产率)。
表2
由此可见,对于有机配体而言,最优选l1,而其它有机配体均在效果上有显著的降低;还可以看出,即便是使用非常类似的l2-l3,其效果也有显著的降低,而l5甚至无法得到产物,这证明有机配体的选择并非是显而易见的。
酸性添加剂的考察
对比例s219-s221:除分别将步骤s2中的对甲苯磺酸一水合物替换为乙酸外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s219-s221。
对比例s222-s224:除分别将步骤s2中的对甲苯磺酸一水合物替换为三氟乙酸外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s222-s224。
对比例s225-s227:除分别将步骤s2中的对甲苯磺酸一水合物替换为苯甲酸外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s225-s227。
对比例s228-s230:除分别将步骤s2中的对甲苯磺酸一水合物替换为甲烷磺酸外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s228-s230。
对比例s231-s233:除分别将步骤s2中的对甲苯磺酸一水合物替换为三氟甲烷磺酸外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s231-s233。
对比例s234-s236:除分别将步骤s2中的对甲苯磺酸一水合物替换为樟脑磺酸外,其它操作均不变,从而重复实施了实施例1-3,顺次得到对比例s234-s236。
结果见下表3(其中产物产率是指步骤s2中化合物(5)的产率)。
表3
nr表示未检测到产物。
由此可见,对于步骤s2中的酸性添加剂而言,稍微的改变都可导致效果上的显著改变,例如当为苯甲酸时,虽然与对甲苯磺酸一水合物高度类似,但仍无法得到产物。这证明酸性添加剂在种类上的选择具有不可预测性和效果上的非显而易见性。
有机溶剂的考察
除分别将有机溶剂甲苯替换为下表4中的其它有机溶剂外,其它操作均不变,从而重复实施了实施例1-3,所使用的有机溶剂、实施例对应关系和产物产率见下表4(其中产物产率是指步骤s2中化合物(5)的产率)。
表4
由此可见,对于有机溶剂而言,最优选甲苯,而其它有机溶剂均在效果上有显著的降低,甚至无法得到产物,这证明有机溶剂的选择并非是显而易见的。
药物活性测试
实验过程如下:
1、配置肉汤,具体为将蛋白胨0.5g、nacl0.25g、可溶性淀粉3g和牛肉膏0.15g用灭菌水定容至50ml;
2、将所得肉汤用0.22μm的滤膜过滤,得到过滤肉汤;
3、向体重18-22g的icr雄鼠(购自于温州医科大学实验动物中心)的鼠腹腔注射5ml肉汤,72h后将icr雄鼠处死,分离得到原代巨噬细胞;
4、将分离的原代细胞离心重悬后接种到六孔板中(每孔5×105个细胞),然后每孔中加入2ml1640完全培养基(gibco),将细胞培养在体积浓度为5%co2、37℃的细胞培养箱中;
5、待细胞贴壁后4-6小时,更换新鲜的1640培养基;
6、细胞贴壁24小时后,再次更换新鲜的1640培养基,每孔加入本发明化合物使其浓度为10nm,随后再加入脂多糖lps(购自sigma-aldrich)使其浓度为0.5mg/ml,继续培养24小时;
7、收集细胞培养基,按照购自ebioscienceinc的小鼠il-6酶联免疫吸附测定(elisa)试剂盒的使用说明操作,测量培养基中的il-6水平,并通过测定总蛋白浓度归一化,结果以对lps的百分比表示(即纵坐标为%)。
结果见附图1,从中可以看出,本发明的化合物k2以及中间体化合物k1均对于il-6具有明显的抑制效果,其中k2的抑制效果要进一步优于k1,从而具有更优异的抗炎活性,在药物领域具有良好的研究前景和应用潜力。
综上所述,本发明提供了一种炎症抑制化合物及其制备方法,所述化合物具有显著的抗炎活性,在药物领域具有良好的研究前景和应用潜力,所述合成方法以简单易得的原料为反应物,经过三步反应而得到该炎症抑制化合物,具有良好的应用前景和研究价值。
应当理解,这些实施例的用途仅用于说明本发明而非意欲限制本发明的保护范围。此外,也应理解,在阅读了本发明的技术内容之后,本领域技术人员可以对本发明作各种改动、修改和/或变型,所有的这些等价形式同样落于本申请所附权利要求书所限定的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!