1-(2,6-氯苯基)-3-(取代嘧啶-4-基)脲类化合物及其制备和应用的制作方法
本发明涉及医药化学技术领域,尤其涉及一种1-(2,6-二氯苯基)-3-(6-(取代苯胺基)嘧啶-4-基)脲类egfr/fgfr双重小分子抑制剂及其制备方法与应用。。
背景技术
受体酪氨酸激酶(receptortyrosinekinases,rtks)是人体中最大的一类酶联受体,人类基因组中已知的rtks共有58种。rtks在细胞生长、增殖、迁移和分化等活动中起着至关重要的作用,从表皮生长因子受体(epidermalgrowthfactorreceptors,egfrs)、成纤维细胞生长因子受体(fibroblastgrowthfactorreceptors,fgfrs)、肝细胞生长因子受体(hepatocytegrowthfactorreceptors,hgfrs)、以及血小板衍生生长因子受体(platelet-derivedgrowthfactorreceptors,pdgfr)等许多rtks发出的信号,已被药理学证明对表达相关蛋白质突变形式的癌症的生存至关重要。靶向这些失调的rtks的酪氨酸激酶抑制剂(tyrosinekinaseinhibitors,tkis),可以给癌症患者带来明显的临床益处。rtks中的egfr和fgfr都已被充分表征为nsclc的重要致癌驱动因素。在过去的十几年中,已有许多egfr-tkis被直接批准用于特定的nsclc患者的临床治疗,例如第一代的egfr-tkis吉非替尼(gefitinib)和厄洛替尼(erlotinib);第二代的egfr-tkis阿法替尼(afatinib);以及第三代的egfr-tkis奥希替尼(osimertinib)。而作为另一重要的抗nsclc的药物作用靶标fgfr,虽说还没有相对应的上市药物,但因为近年来,有大量的体外研究和独立实验表明,在nsclc中存在着fgfr1的基因扩增和fgfr2的过表达。且在一项对59个nsclc患者的定量rt-pcr实验中发现,nsclc患者的肿瘤组织fgfr1的转录活性是癌旁组织的两倍多。因此,越来越多的fgfr-tkis也正被报道用于治疗nsclc的探索研究。
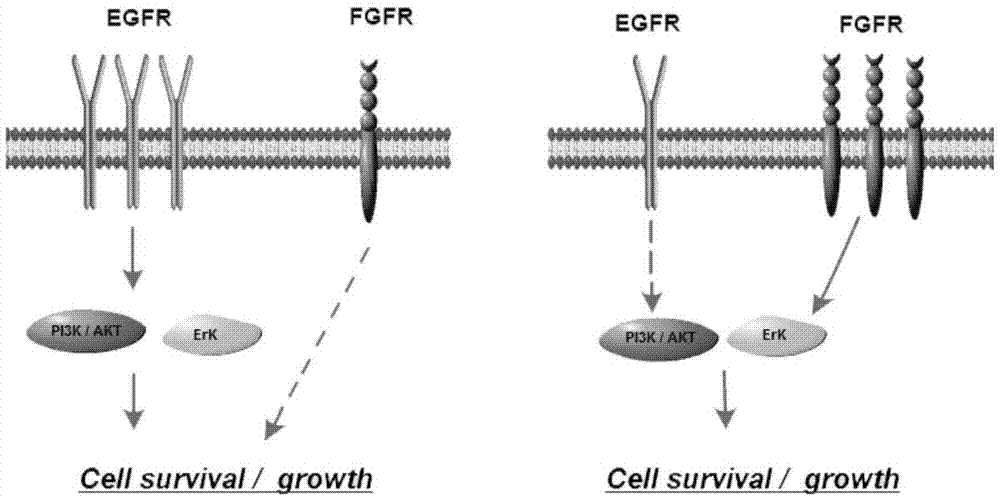
然而,尽管已有多种化学型的小分子抑制剂作为egfr-tkis和fgfr-tkis进行了抗nsclc的广泛研究,但迄今为止,还没有真正可以作为两种激酶受体的双重小分子抑制剂的相关报道。已经证明,临床使用egfr-tkis如吉非替尼或阿法替尼治疗后,会以负反馈的方式增加nsclc细胞株的fgfr1的扩增而产生耐药(原理见图1),而通过基因沉默方式敲除fgfr1,或是同fgfr-tkis进行联合用药处理,可以有效抑制耐药株的形成。由此可见,对egfr靶向抑制而导致的fgfr负反馈活化,即旁路激活,也是egfr-tkis产生耐药抵抗的重要原因之一;并且,随着进行单细胞测序的能力提高,已有报道同时携带突变型egfr和fgfr的致癌等位基因的肿瘤。考虑到,不同类别的药物联合使用,往往存在患者用药不便、药物相互作用或配伍禁忌,结果出现一些毒副作用或不良反应。所以,egfr与fgfr的双重抑制剂的研发特别是对于nsclc及其耐药治疗,具有迫切的临床需要和发展前景。
技术实现要素:
本发明提供了一种1-(2,6-氯苯基)-3-(取代嘧啶-4-基)脲类化合物及其制备和应用,这类化合物具有较好的抗肿瘤活性,并可以在一定程度上防止耐药性的产生,并且毒副作用较小。
本发明采用如下技术方案:
本发明的1-(2,6-二氯苯基)-3-(6-(取代苯胺基)嘧啶-4-基)脲类egfr/fgfr双重小分子抑制剂的化学结构如下:
其中,r的取代基表1-1所示:
表1-1:f系列化合物的结构
其中所述的化合物1-(2,6-二氯苯基)-3-(6-((3,5-二甲氧基苯基)氨基)嘧啶-4-基)脲(f1),化学结构如下:
其中所述的化合物1-(2,6-二氯苯基)-3-(6-((2,5-二甲氧基苯基)氨基)嘧啶-4-基)脲(f2),化学结构如下:
其中所述的化合物1-(2,6-二氯苯基)-3-(6-((3-氟-5-甲基苯基)氨基)嘧啶-4-基)脲(f8),化学结构如下:
本发明还提供了一种制备1-(2,6-二氯苯基)-3-(6-(取代苯胺基)嘧啶-4-基)脲类egfr/fgfr双重小分子抑制剂,其特征在于:第一步中间产物的合成:
步骤一:取一干燥的三口反应瓶,放入磁石。加入4-氨基-6-氯嘧啶(1eq),ki(0.5eq),并用的无水乙醇(35ml)溶解。在磁力搅拌器上,搅拌加热10min后,加入三氟乙酸(200ml)。活化。大约1h后,加入无水乙醇(15ml)溶解的取代苯胺(0.8eq)反应。注意,要以滴加的方式加进去,以达到长时过量反应的效果,滴加时间控制在1h左右。
步骤二:用tlc法,检测反应进程和反应效果。一般是反应36h后,反应几乎完全。反应完全后,先旋干溶剂无水乙醇,然后加入一定量的乙酸乙酯溶解。先用适量的20%的碳酸氢钠溶液除酸,后再加入一定量的50%的氯化钠溶液萃取分层,收集有机层并旋干至剩一定量,加入无水硫酸钠适量,除水过夜。
步骤三:抽滤,制砂,称取原料总和15到20倍的柱层硅胶粉装柱,过柱收集产物点。一般先是用石油醚:乙酸乙酯=2:1过出第一个点(苯胺类点),石油醚:乙酸乙酯=1:1过出第二个点(嘧啶类点),乙酸乙酯或甲醇冲出产物点。收集旋干产物点,放烘箱干燥,打质谱及核磁,验证。
本发明还提供了一种制备1-(2,6-二氯苯基)-3-(6-(取代苯胺基)嘧啶-4-基)脲类egfr/fgfr双重小分子抑制剂,其特征在于:第二步中间产物的合成:
步骤一:取一干燥的三口反应瓶,加入磁石。称取三光气(0.5eq),并用二氯甲烷(20ml)溶解,超声使其充分。于0℃下,缓慢滴加二氯甲烷(10ml)溶解的2,6-二氯苯胺(1eq),每一分钟一滴,滴加时间控制在0.5h左右。滴毕,记得加2~3滴的三乙胺。加热回流反应3~6h,反应一般会完全。减压浓缩至干,剩余物用乙酸乙酯溶解,依次用10%的硫酸氢钾溶液除碱、20%的碳酸氢钠溶液除酸和50%的氯化钠溶液洗涤除无机相,收集有机相并旋干,经无水硫酸钠除水干燥过夜后,抽滤,制砂。
步骤二:制砂,装柱,过出2,6-二氯苯胺后,分离提纯异氰酸酯。送去打质谱和核磁,验证。
本发明还提供了一种制备1-(2,6-二氯苯基)-3-(6-(取代苯胺基)嘧啶-4-基)脲类egfr/fgfr双重小分子抑制剂,其特征在于:第三步目标产物的合成:
步骤一:取一干燥的三口反应瓶,加入磁石。称取的嘧啶苯胺(1eq),异氰酸酯(1.2eq),并用甲苯(10ml)溶解,于70~80℃加热回流反应8~10h,基本反应完全,tlc检测。冷却至室温,滤饼用甲苯洗涤2~3次,刮下后用乙酸乙酯溶解,20%的碳酸氢钠溶液洗涤除酸,再加入50%的氯化钠溶液萃取分层。收集并旋干有机层。
步骤二:点板检测纯度,送去打质谱及核磁,验证。
本发明的1-(2,6-二氯苯基)-3-(6-(取代苯胺基)嘧啶-4-基)脲类衍生物可以用于肿瘤的治疗。
本发明的1-(2,6-二氯苯基)-3-(6-(取代苯胺基)嘧啶-4-基)脲类衍生物表现出一定的抗肿瘤活性。根据抗肿瘤活性测试结果,化合物f1、f2和f8对五个非小细胞肺癌细胞系,都表现出了一定的抑制活性;其中,活性最好的是化合物f2。化合物f2作用于5个癌细胞的ic50值分别为2.15±0.32μm,1.34±0.25μm,3.03±0.42μm,2.53±0.37μm,1.98±0.29μm。此外,化合物f2的激酶筛选结果显示,化合物f2能直接对激酶受体egfr和fgfr产生抑制效果,且有一定程度的选择性。因而,化合物f2是一个有效的egfr/fgfr抑制剂。
附图说明
图1为fgfr1的激活介导egfr-tkis获得性耐药的产生原理示意图。
图2为实施例2得到的抗肿瘤细胞试验的结果。
具体实施方式
下面的实施例是对本发明的进一步详细描述。
实施例1化合物的合成
1.1化合物的具体合成路线如下所示:
1.2合成步骤
a.第一步中间产物的合成:
步骤一:取一干燥的三口反应瓶,放入磁石。加入4-氨基-6-氯嘧啶(1eq),ki(0.5eq),并用的无水乙醇(35ml)溶解。在磁力搅拌器上,搅拌加热10min后,加入三氟乙酸(200ml)。活化。大约1h后,加入无水乙醇(15ml)溶解的取代苯胺(0.8eq)反应。注意,要以滴加的方式加进去,以达到长时过量反应的效果,滴加时间控制在1h左右。
步骤二:用tlc法,检测反应进程和反应效果。一般是反应36h后,反应几乎完全。反应完全后,先旋干溶剂无水乙醇,然后加入一定量的乙酸乙酯溶解。先用适量的20%的碳酸氢钠溶液除酸,后再加入一定量的50%的氯化钠溶液萃取分层,收集有机层并旋干至剩一定量,加入无水硫酸钠适量,除水过夜。
步骤三:抽滤,制砂,称取原料总和15到20倍的柱层硅胶粉装柱,过柱收集产物点。一般先是用石油醚:乙酸乙酯=2:1过出第一个点(苯胺类点),石油醚:乙酸乙酯=1:1过出第二个点(嘧啶类点),乙酸乙酯或甲醇冲出产物点。收集旋干产物点,放烘箱干燥,打质谱及核磁,验证。
b.第二步中间产物的合成:
步骤一:取一干燥的三口反应瓶,加入磁石。称取三光气(0.5eq),并用二氯甲烷(20ml)溶解,超声使其充分。于0℃下,缓慢滴加二氯甲烷(10ml)溶解的2,6-二氯苯胺(1eq),每一分钟一滴,滴加时间控制在0.5h左右。滴毕,记得加2~3滴的三乙胺。加热回流反应3~6h,反应一般会完全。减压浓缩至干,剩余物用乙酸乙酯溶解,依次用10%的硫酸氢钾溶液除碱、20%的碳酸氢钠溶液除酸和50%的氯化钠溶液洗涤除无机相,收集有机相并旋干,经无水硫酸钠除水干燥过夜后,抽滤,制砂。
步骤二:制砂,装柱,过出2,6-二氯苯胺后,分离提纯异氰酸酯。送去打质谱和核磁,验证。
c.第三步目标产物的合成:
步骤一:取一干燥的三口反应瓶,加入磁石。称取的嘧啶苯胺(1eq),异氰酸酯(1.2eq),并用甲苯(10ml)溶解,于70~80℃加热回流反应8~10h,基本反应完全,tlc检测。冷却至室温,滤饼用甲苯洗涤2~3次,刮下后用乙酸乙酯溶解,20%的碳酸氢钠溶液洗涤除酸,再加入50%的氯化钠溶液萃取分层。收集并旋干有机层。
步骤二:点板检测纯度,送去打质谱及核磁,验证。
1.3实验结果
合成的所有目标化合物结构如上表1-1所示;合成的包括活性化合物在内的部分目标化合物的ms、1hnmr和13cnmr等理化数据如下:
1-(2,6-dichlorophenyl)-3-(6-((3,5-dimethoxyphenyl)amino)pyrimidin-4-yl)urea(f1).
yellowpowder,yield:45.6%;mp/℃:201.7~202.3;esi-ms[m+na]+:434.12;1hnmr(600mhz,cdcl3-d6)δ(ppm):8.389(s,1h,-nh-),7.371(s,1h,2-pyrimidine-h),7.358(s,3h,ar-h),7.199(s,1h,ar-h),7.187(s,1h,ar-h),6.503(s,1h,ar-h),6.302(s,1h,5-pyrimidine-h),3.792(s,6h,-och3).13cnmr(150mhz,dmso-d6)δ(ppm):161.674,161.144,157.705,152.309,142.200,134.182,133.100,129.388,129.114,128.785,98.892,94.786,90.830,79.567,55.644.
1-(2,6-dichlorophenyl)-3-(6-((2,5-dimethoxyphenyl)amino)pyrimidin-4-yl)urea(f2).
taupepowder,yield:44.3%;mp/℃:195.4~196.1;esi-ms[m+na]+:433.92;1hnmr(600mhz,cdcl3-d6)δ(ppm):8.508(s,1h,-nh-),8.439(s,1h,2-pyrimidine-h),7.781(s,1h,ar-h),7.377(d,j=7.8hz,1h,ar-h),7.364(d,j=7.8hz,1h,ar-h),7.195(m,1h,ar-h),7.180(s,1h,ar-h),6.823(m,1h,ar-h),6.586(s,1h,5-pyrimidine-h),3.822(s,3h,-och3),3.786(s,3h,-och3).13cnmr(150mhz,dmso-d6)δ(ppm):162.343,157.657,153.576,152.030,146.255,134.196,133.001,129.120,129.086,112.977,111.051,109.206,91.311,91.180,56.700,55.987.
1-(6-((4-bromo-2-methoxyphenyl)amino)pyrimidin-4-yl)-3-(2,6-dichlorophenyl)urea(f4).
taupepowder,yield:38.3%;mp/℃:203.3~204.0;esi-ms[m+na]+:482.00;1hnmr(600mhz,cdcl3-d6)δ(ppm):8.523(s,1h,-nh-),8.436(s,1h,2-pyrimidine-h),7.412(d,j=7.8hz,1h,ar-h),7.399(d,j=7.8hz,1h,ar-h),7.189(m,4h,ar-h),6.822(s,1h,5-pyrimidine-h),3.911(s,3h,-och3).13cnmr(150mhz,dmso-d6)δ(ppm):162.304,162.147,158.135,157.707,157.580,157.412,152.985,152.302,134.204,129.436,129.127,129.026,127.939,126.036,123.588,115.347.
1-(2,6-dichlorophenyl)-3-(6-((3-fluoro-5-methylphenyl)amino)pyrimidin-4-yl)urea(f8).
whitepowder,yield:40.9%;mp/℃:229.8~231.0;esi-ms[m+na]+:406.08;1hnmr(600mhz,cdcl3-d6)δ(ppm):8.409(s,1h,-nh-),7.381(d,j=7.8hz,1h,ar-h),7.368(d,j=7.8hz,1h,ar-h),7.178(d,j=7.8hz,1h,ar-h),7.165(d,j=7.8hz,1h,ar-h),7.025(s,1h,2-pyrimidine-h),6.919(s,1h,ar-h),6.722(s,1h,ar-h),6.708(s,1h,5-pyrimidine-h),2.364(s,3h,-ch3).13cnmr(150mhz,dmso-d6)δ(ppm):163.611,162.023,161.665,161.526,158.124,157.979,157.775,152.278,134.191,133.047,129.473,129.157,116.306,21.781.
1-(2,6-dichlorophenyl)-3-(6-((2-methoxy-5-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)urea(f11).
whitepowder,yield:34.3%;mp/℃:239.1~240.2;esi-ms[m+na]+:472.08;1hnmr(600mhz,cdcl3-d6)δ(ppm):8.483(s,1h,-nh-),8.481(s,1h,2-pyrimidine-h),7.399(m,4h,ar-h),7.218(d,j=8.4hz,1h,ar-h),7.204(d,j=8.4hz,1h,ar-h),6.987(s,1h,5-pyrimidine-h),3.972(s,3h,-och3).13cnmr(150mhz,dmso-d6)δ(ppm):162.211,158.020,157.667,153.005,134.205,129.331,129.129,121.652,121.225,119.944,112.211,92.328,56.682.
1-(6-((3-(tert-butyl)phenyl)amino)pyrimidin-4-yl)-3-(2,6-dichlorophenyl)urea(f15).
whitepowder,yield:39.8%;mp/℃:222.1~222.9;esi-ms[m+na]+:430.21;1hnmr(600mhz,cdcl3-d6)δ(ppm):8.659(s,1h,-nh-),8.471(m,2h,ar-h),8.394(s,1h,2-pyrimidine-h),7.402(d,j=8.4hz,1h,ar-h),7.388(d,j=8.4hz,1h,ar-h),7.183(m,3h,ar-h),7.001(s,1h,5-pyrimidine-h),1.342(s,9h,-ch3).13cnmr(150mhz,dmso-d6)δ(ppm):161.974,158.010,157.590,152.257,151.913,140.128,128.987,120.146,118.115,117.844,35.026,31.735.
1-(6-(benzo[d][1,3]dioxol-5-ylamino)pyrimidin-4-yl)-3-(2,6-dichlorophenyl)urea(f17).
taupepowder,yield:31.1%;mp/℃:260.4~261.2;esi-ms[m+na]+:417.98;1hnmr(600mhz,cdcl3-d6)δ(ppm):8.328(s,1h,-nh-),7.410(m,4h,ar-h+2-pyrimidine-h),7.195(t,j=8.4hz,1h,ar-h),6.851(d,j=8.4hz,1h,ar-h),6.837(d,j=8.4hz,1h,ar-h),6.779(s,1h,5-pyrimidine-h),6.037(s,2h,ch2).13cnmr(150mhz,dmso-d6)δ(ppm):161.918,158.049,157.762,152.350,147.765,143.328,134.635,134.200,133.112,129.426,129.132,114.147,108.694,103.597,101.564,89.888,72.872,60.851.
本发明所合成目标化合物的性状及其溶解性如下:
目标化合物产率普遍较高。化合物f1为黄色固体;化合物f2,f4,f7和f17,均为灰褐色固体;化合物f3,f5,f8-9,f11,f13-14,f16,均为白色固体;化合物f10,f12和f18,均为黑色固体;化合物f6为淡绿色固体。化合物易溶于乙酸乙酯、乙腈、二氯甲烷、dmso、dmf;微溶于石油醚、甲醇、乙醇;不溶于甲苯。
本发明合成的目标化合物,在ms谱图中均显示了[m+1]+峰,且信号较强,部分化合物存在着同位素峰。1h-nmr谱图结果显示,所有目标化合物的氢信号,以及其化学位移,在图谱上都能清晰的看出。以dmso-d6为溶剂时,核磁氢谱数据显示完全,即化合物氢的理论个数,与核磁氢谱图上氢的个数相吻合;而以cdcl3-d6为溶剂时,大多数的目标化合物的核磁氢谱数据显示不完全,核磁氢谱图上通常没有脲基胺上的两个氢。13c-nmr谱图结果显示,目标化合物碳峰位移及数目基本上与理论数据相符。
实施例2化合物抗肿瘤细胞活性
2.1mtt法测试化合物抗肿瘤活性
本实验采用mtt法。所选的正常肺细胞为beas-2b细胞;所选的五个非小细胞肺癌细胞则包括肺腺癌细胞a549(wtegfr)、肺腺癌细胞pc-9(egfrd746-750)、肺鳞癌细胞h520(fgframplification)、大细胞肺癌h1581(fgframplification)和肺鳞癌细胞h226(fgframplification/overexpressedegfr)。选取对数生长的上述细胞,将这些细胞消化,收集,并用细胞计数板进行计数。接着,将记过数的细胞,稀释至合适的浓度(5*10^4个/ml~8*10^4个/ml),按每孔100μl将稀释的细胞悬液用排枪加入到96孔板中进行培养,并记得在同一块孔板上设置只含培养基的空白对照孔;铺板过夜培养后,更换为新鲜培养基,每孔加入一系列浓度梯度稀释的受试目标化合物和阳性药物bgj398和g,等药物作用72小时后,检测细胞的存活率;将20μl的mtt检测液,加入到96孔板的每个孔中,后将96孔板放置在37℃培养箱中,孵育四小时。去上清,并加入150μl的dmso,溶解mtt甲臢沉淀。最后以酶标仪检测紫外吸收波长在490nm处的每孔的吸光值,并进行换算,计算出相应的细胞存活率,抑制率或ic50值等。本实验需进行至少三次重复实验,减小实验误差。
2.2实验结果
试验结果如图2所示,如图2a所示,在10μm的浓度下,所有受试目标化合物,对应的细胞存活率均在95%以上;而两药联用的阳性对照组,对应的细胞存活率较低,为75%。图2b显示的是受试化合物对a549细胞的抑制效果,其中抑制率超过50%的化合物为f1~f2、f4~f14、f17;图2c所示为受试化合物对pc-9细胞的抑制效果,其中抑制率超过50%的为化合物f1~f15、f17~f18,化合物f2、f7、f9、q4的抑制效果阳性药g强;受试化合物对h520细胞的抑制效果,如图2d所示,化合物f1~f2、f8的抑制率都超过50%;受试化合物对h1581细胞的抑制效果,如图2e所示,化合物f1~f2、f5~f6、f8~f9、f11、f13、f15~f16的抑制率都超过50%,其中化合物f2的抑制效果与阳性药bgj398相当;而对h226细胞的抑制效果,则如图2f所示,化合物f1~f2、f5~f6、f8、f10~f11、f13、f17的抑制率都超过50%,其中最好的化合物是f2,抑制效果好于阳性药g或bgj398的单药使用,但弱于阳性药g和bgj398的两药联用。
实例3化合物f1、f2和f8对五种肿瘤细胞的ic50实验
3.1mtt法测试化合物的ic50值
根据所有目标化合物对正常肺细胞的毒性作用结果,以及对五种nsclc细胞系的抑制作用结果,我们得到了对五种nsclc细胞系都起抑制效果的化合物f1、f2和f8。我们选择这三个化合物,进一步地去做ic50实验。先为这两个化合物都设定六种浓度(20μm,10μm,1.0μm,0.50μm,0.10μm和0.01μm)。然后,分别用不同浓度的这两个化合物处理五种非小细胞肺癌细胞系,包括a549细胞、pc-9细胞、h520细胞、h1581细胞和h226细胞。获得光密度(od)值,计算抑制率,并通过graphpadprism5软件计算不同化合物的ic50值。
3.2实验结果
对a549细胞、pc-9细胞、h520细胞、h1581细胞和h226细胞这五个癌细胞的ic50值,化合物f1测得的结果分别为13.32±1.53μm、3.18±0.68μm、10.46±1.34μm、5.32±0.75μm和16.85±2.09μm;而化合物f2所测得结果则分别为2.15±0.32μm、1.34±0.25μm、3.03±0.42μm、2.53±0.37μm和1.98±0.29μm;化合物f8测得结果分别为18.64±1.98μm、2.79±0.34μm、3.78±0.56μm、4.78±0.68μm和2.96±0.36μm。该系列中活性最好的是化合物f2。
实例4化合物f2的体外激酶活性初筛
4.1msa法测化合物f2对激酶的抑制作用
实验中使用的是calipermobilityshiftassay方法,该方法是以微流体芯片技术的迁移检测技术为核心的检测平台。实验步骤如下:先配制1×kinasebuffer。再进行化合物浓度梯度的配制:受试化合物f2测试浓度为10μm起始,4倍稀释10个浓度,复孔测试,在96孔板中梯度稀释成100倍终浓度的10个不同浓度的溶液。然后用1×kinasebuffer将各浓度的化合物进一步稀释成5倍终浓度的中间稀释溶液。将配制好的化合物溶液各取5μl分别加入384孔板的化合物孔,每个浓度单孔测试;阴性对照孔和阳性对照孔中加5μl的5%dmso。用1×kinasebuffer配制2.5倍终浓度的激酶溶液。在化合物孔和阳性对照孔分别加10μl的2.5倍终浓度的激酶溶液;在阴性对照孔中加10μl的1×kinasebuffer。1000rpm离心30秒,振荡混匀后室温孵育10分钟。用1×kinasebuffer配制2.5倍终浓度的atp和kinasesubstrate的混合溶液。加入10μl的2.5倍终浓度的atp和底物的混合溶液,起始反应。将384孔板1000rpm离心30秒,振荡混匀后28℃分别孵育相应的时间。加入30μl终止检测液停止激酶反应,1000rpm离心30秒,振荡混匀。最后,在caliper上测试收集数据,计算公式:inhibition=(conversion%_max-conversion%_sample)/(conversion%_max-conversion%_min)*100。其中:conversion%_sample是样品的转化率读数;conversion%_min:阴性对照孔均值,代表没有酶活孔的转化率读数;conversion%_max:阳性对照孔比值均值,代表没有化合物抑制孔的转化率读数。
4.2实验结果
表4-110μm化合物f2的激酶抑制率
筛选结果显示,化合物f2对egfr(wt)、egfr(d746-750)、egfrt790m、egfrl858r、fgfr1和fgfr2的抑制率分别为92.81%、95.47%、70.98%、98.45%、75.17%和88.51%,抑制效果明显。然而,对于其他激酶受体如jak、akt和met等的抑制作用比较弱,抑制率最高的仅能达到28.01%。从该实验结果,我们可以初步断定的是,化合物f2能直接对激酶受体egfr和fgfr产生明显的抑制效果,且化合物f2对激酶受体egfr和fgfr具有一定程度上的选择性。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种咪康唑药物中间体α‑(2,4‑二氯苯基)‑β‑(1‑咪唑基)乙醇的合成方法与制造工艺
- 一种烯丙尼定药物中间体2‑(N‑烯丙基‑N‑2,6‑二氯苯基)氨基咪唑啉的合成方法与制造工艺
- 一种啶酰菌胺中间体2‑(4‑氯苯基) 硝基苯的制备方法与制造工艺
- 一种咪康唑药物中间体2,4,‑二氯苯基氯甲基甲酮的合成方法与制造工艺
- 一种阿立哌唑口服用液体干混悬剂及其制备方法与制造工艺
- 一种铑‑双磷配体四氟化硼盐及制备方法与制造工艺
- 用于制备AZD5363的方法和在其中使用的新颖中间体与制造工艺
- 用于治疗ADHD的达斯曲林(DASOTRALINE)剂量和方法与制造工艺
- 用于治疗ADHD的方法和达斯曲林(DASOTRALINE)组合物与制造工艺
- 药物活性物质的固体形式的制造方法与工艺