外电场下盐交联聚乙烯分子结构及分析外电场下盐交联聚乙烯分子结构构建的方法与流程
本发明外电场下盐交联聚乙烯分子结构的变化规律,涉及高电压输电电缆绝缘老化领域。
背景技术:
聚乙烯和交联聚乙烯以其优异的电气性能、力学性能及工艺性能,已成为电力电缆最主要的绝缘材料。随着高压直流输电技术的发展,以及大规模远距离输电和新能源消纳的要求,聚乙烯及交联聚乙烯正逐渐被应用于高压直流电缆绝缘中。当聚合物在高电场的长期作用下,这些杂质解离容易导致聚合物内部空间电荷的积累,可使局部电场强度发生严重的畸变。畸变场强在绝缘材料内部形成放电,引发水树枝和化学树枝,它们都将转化为电树枝而导致绝缘击穿,树枝状老化是引起交联聚乙烯电缆绝缘击穿的关键因素。因此,外电场研究对聚合物绝缘性能有重要意义。
目前针对聚合物绝缘性能的实验研究主要集中于温度和交联副产物对空间电荷分布的作用,不同温度和局部气压对电树枝生长的影响,双极性直流电场对水树枝生长和取向研究,纳米材料填充改性绝缘电缆。而随着分子模拟技术的发展,其也越来越多的应用于高电压与绝缘技术领域,如构建电树枝生长长度和分形维数随时间变化的模型方程以及聚乙烯的完全物态方程,电场和温度对聚合物空间电荷陷阱深度的研究,预电力对聚合物击穿性能的影响,电缆内导电屏蔽层与绝缘层的界面相容性分析。但是从分子模拟的微观角度分析xlpe在外电场下分子结构变化和特性的研究还鲜见报道。
技术实现要素:
有鉴于此,本发明所要解决的技术问题是:针对交联聚乙烯绝缘电缆中的盐交联聚乙烯在外电场下的微观特性,使用密度泛函理论研究外电场下盐交联聚乙烯分子结构的变化规律。与现在常用的实验方法相比,具有无损耗、节约成本、简单易行等优点,揭示微观结构和外电场之间的关系,为今后实验研究与微观研究提供理论基础和数据依据。
本发明采取的技术方案:
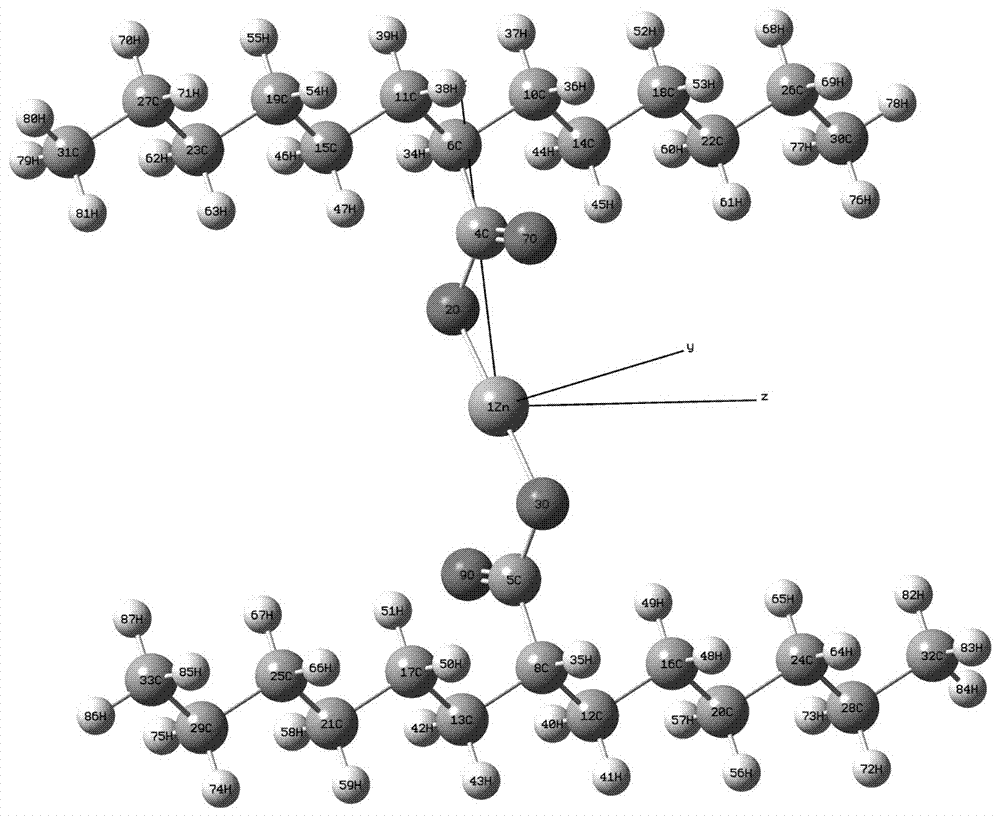
外电场下盐交联聚乙烯分子结构的变化规律,首先利用分子模拟软件构建羧酸盐(-coozn2+ooc-)结构盐桥的交联聚乙烯分子模型,聚乙烯单链使用10-15个烷烃结构(所述的聚乙烯单链为13个烷烃组成的链烷烃结构)。使用明尼苏达泛函优化初始分子结构,对优化之后的分子结构施加外电场并对其进行结构优化,得到不同外电场下分子结构的稳定结构。分析不同外电场作用下盐交联聚乙烯分子结构和能量变化,外电场对前线轨道的能级和成分的影响,原子之间的键级、断键和红光光谱的变化。上述所述的外电场下盐交联聚乙烯分子,该分子结构中交联聚乙烯分子为h型结构,聚乙烯链为空间网状结构,其结构式如下:
进一步的在外电场0.010-0.020a.u.的优化作用下,交联聚乙烯分子为x型结构,聚乙烯链为线性结构,交联处的四个氧原子与锌原子形成四面体的四配位结构,d2d点群对称性,其结构式如下:
上述结构的演变过程中,在外电场0.010-0.020a.u.的逐渐增加的条件下,沿电场方向聚乙烯链端为亲电反应活性,c-c键断裂,形成甲基碳负离子;逆电场方向聚乙烯链端为亲核反应活性,c-h键断裂形成h正离子。
在外电场0.010-0.020a.u.的逐渐增加的条件下,两条聚乙烯链之间的链端的距离逐渐减小,二面夹角逐渐减小。
本发明所述的外电场下盐交联聚乙烯分子的构建方法,包括如下方法:
步骤1:在分子模拟软件内构建羧酸盐-coozn2+ooc-结构盐桥的交联聚乙烯分子模型,聚乙烯单链为10-15个烷烃结构(所述的聚乙烯单链为13个烷烃组成的链烷烃结构),运用m06-2x/gen,zn原子使用def2-tzvp基组,c、h、o原子使用6-31g(d)基组,对交联聚乙烯分子进行优化,获得分子能量最低时的优化结构;
步骤2:运用m06-2x/gen,zn原子使用def2-tzvp基组,c、h、o原子使用6-31g(d)基组,方法对步骤1优化之后的分子结构施加外电场并对其进行结构优化,得到不同外电场下分子结构的稳定结构,外电场强度以0.002a.u.为步长对分子施加0-0.020a.u.的电偶极场;
步骤3:从不同外电场作用下,分析分子结构中两条聚乙烯链之间的夹角和链端的距离分别随外电场的变化,分析分子在外电场下的结构变化,分子总能量、偶极矩、极化率随外电场的变化趋势;
步骤4:分析不同外电场下交联聚乙烯分子的前线轨道的能级和分布的变化,确定分子结构的反应活性位点,并分析不同外电场下分子前线轨道的主要成分。
本发明外电场下盐交联聚乙烯分子结构的变化规律,技术效果如下:
1:本发明基于物质分子结构,利用计算化学软件,使用密度泛函理论优化计算分子在不同外电场下的基态结构,准确判定不同外电场作用下盐交联聚乙烯分子结构和能量变化,外电场对前线轨道的能级和成分的影响,原子之间的键级、断键和红光光谱的变化。
2:与现在常用的实验方法相比,具有无损耗、节约成本、简单易行等优点,揭示交联聚乙烯分子微观结构和外电场之间的关系,为今后实验研究与微观研究提供理论基础和数据依据。
附图说明
图1为盐交联聚乙烯初始分子模型。
图2为盐交联聚乙烯优化分子模型。
图3为聚乙烯链端部的距离r随外电场的变化。
图4为聚乙烯链端的二面角d随外电场的变化。
图5为总能量随外电场的变化。
图6为偶极矩随外电场的变化。
图7为极化率随外电场的变化。
图8为前线轨道能级随外电场的变化。
图9为能隙eg随外电场的变化。
图10为外电场下前线轨道分布(a)、(b)和(c)分别是电场为0、0.010和0.020a.u.的homo;(d)、(e)和(f)分别是电场为0、0.010和0.020a.u.的lumo。
图11为外电场下mbo(a)链端的c-c键的mbo值随外电场的变化。
图12为外电场下mbo(a)(b)链端的c-h键的mbo值随外电场的变化。
图13为盐交联聚乙烯e分子在不同外电场下的ir光谱。
具体实施方式
外电场下盐交联聚乙烯分子结构的变化规律,首先利用分子模拟软件构建羧酸盐(-coozn2+ooc-)结构盐桥的交联聚乙烯分子模型,本发明的优选方案中聚乙烯单链使用13个烷烃结构。使用明尼苏达泛函优化初始分子结构,对优化之后的分子结构施加外电场并对其进行结构优化,得到不同外电场下分子结构的稳定结构。分析不同外电场作用下盐交联聚乙烯分子结构和能量变化,外电场对前线轨道的能级和成分的影响,原子之间的键级、断键和红光光谱的变化。
外电场下盐交联聚乙烯分子结构的变化规律,包括以下步骤:
步骤1:在分子模拟软件内构建羧酸盐(-coozn2+ooc-)结构盐桥的交联聚乙烯分子模型,聚乙烯单链使用13个烷烃结构,如图1所示。运用m06-2x/gen(zn原子使用def2-tzvp基组,c、h、o原子使用6-31g(d)基组)方法对交联聚乙烯分子进行优化,获得分子能量最低时的优化结构。交联聚乙烯分子由初始的h型结构变成了x型结构,聚乙烯链稍微向外侧弯曲,交联处的四个氧原子与锌原子形成四面体的四配位结构(d2d点群对称性),如图2所示。
步骤2:运用m06-2x/gen(zn原子使用def2-tzvp基组,c、h、o原子使用6-31g(d)基组)方法对步骤1优化之后的分子结构施加外电场并对其进行结构优化,得到不同外电场下分子结构的稳定结构。
以0.002a.u.为步长对分子施加0-0.020a.u.的电偶极场并对其进行基态几何结构优化。
步骤3:从不同外电场下分子结构的数据中可知,两条聚乙烯链之间的夹角和链端的距离分别随外电场的变化,分析分子在外电场下的结构变化,分子总能量、偶极矩、极化率随外电场的变化趋势,其计算结果如表1所示。
表1不同外场下分子基态的几何结构、能量、偶极矩和极化率
r(30,32)和r(31,33)分别表示两条聚乙烯链端部30c与32c之间的距离和31c与33c之间的距离,随着外电场的增大,r(30,32)和r(31,33)都减小,但逆电场侧的r(31,33)比沿电场侧的r(30,32)减少的更多,如图3所示。d(30,6,8,32)和d(31,6,8,33)表示两条聚乙烯链的二面角,随着外电场的增大,它们的二面角都在逐渐减小,d(30,6,8,32)从初始的71°变成13°,而d(31,6,8,33)从初始的69°变成6°。聚乙烯链在外电场下发生取向效果,从空间网状结构逐渐变成线性结构,且沿电场方向取向效果更明显,如图4所示。随着外电场的增大,体系总能量逐渐降低,分子体系能量随外电场的变化,如图5所示。分子的偶极矩随着外电场的增大逐渐增大,如图6所示。极化率随着外电场的增大单调递增,且变化越来越快,如图7所示。模拟计算中当电场超过0.0217a.u.时优化不收敛,计算的临界击穿场强是0.0217a.u.(11.16gv/m)。
步骤4:运用相同的方法计算分子在不同外电场下的最高占据轨道(homo)能级eh、最低空轨道(lumo)能级el和最低空轨道与最高占据轨道的能级之差—能隙eg,计算结果如表2所示。
表2不同外电场下分子的前线轨道能级变化
前线轨道能级和能隙的变化如图8、图9所示。外电场在0.011a.u.(5.66gv/m)之前,电场对能隙eg的影响主要是由homo轨道贡献的,lumo轨道的能级变化很小,分子亲核性增大,亲电性不变。外电场在0.011a.u.之后,电场对能隙eg的影响分别由homo和lumo轨道贡献,分子亲核性和亲电性都增加。在外电场为0、0.010和0.020a.u.时候分子的homo和lumo轨道,如图10所示。同时使用hirshfeld方法分析分子轨道中各主要原子的贡献,如表3所示。当无外电场的时候,homo轨道主要是由两条聚乙烯链上的c原子贡献,而lumo轨道集中于交联盐桥,有48%是由zn原子贡献,四个氧原子贡献7%左右;当外电场为0.010a.u.的时候,homo轨道主要是由逆电场方向的聚乙烯链最外侧的三个c原子分别贡献20%左右和一个h原子贡献14%,而lumo轨道中zn原子的贡献降低了2%,2c和3c的贡献降低了大概3%,7c和9c的贡献增加了大概2%;当外电场为0.020a.u.的时候,homo轨道超过60%的贡献是由逆电场方向的聚乙烯链最外侧的甲基提供,其中c起主要作用,表示c更容易失去电子,而lumo轨道近80%的贡献是由沿电场方向的聚乙烯链最外侧的甲基提供,甲基更容易得到电子。
表3不同外电场下分子前线轨道的主要成分
步骤5:mayerbondorder(mbo)是由电子云密度和重叠矩阵计算所得,它能直观地描述原子间同类键的强弱。mbo的值越小,表示该键越弱,键能越小越容易断裂,分析位于聚乙烯链端的c-c键和c-h键随外电场变化时mbo值的变化趋势,判断其受到攻击会发生什么反应,mbo值随外电场的变化如表4所示。
表4c-c键和c-h键的mbo随外电场的变化
c-c和c-h键的mbo值随电场的变化如图11所示。c-c键的mbo值随外电场的增大而逐渐降低,键能逐渐降低,但是处于正半轴的c-c键mbo值降低的更快,表示正半轴的c-c键最先断裂,由于沿电场方向的聚乙烯链端表现出亲电反应活性,当c-c键断裂的时候,甲基团很可能夺得一个电子,形成甲基碳负离子。随着外电场的增大,处于正半轴的c-h键的mbo值逐渐升高,键能逐渐升高;而处于负半轴的c-h键的mbo值逐渐降低,键能逐渐降低,表示处于负半轴的c-h键最先断裂,由于逆电场方向的聚乙烯链端表现出亲核反应活性,氢原子很可能失去一个电子,形成h正离子。
步骤6:在外电场逐渐增大的情况下,使用与优化相同的方法对图2所示模型的分子进行频率计算,分别得到了它在外电场为0、0.010、0.020a.u.时的红外(infrared,ir)光谱,如图12所示。图中列出了无外电场时的11个特征峰,波数426cm-1和492cm-1都是o-zn-o的对称伸缩振动,当波数是426cm-1时出现了红移现象,其键能减小;波数是492cm-1时出现了蓝移现象,其键能增大。578cm-1的吸收峰归属于交联处c-c-o的变角震动,出现了明显的蓝移现象,其键能在电场下逐渐变大。696cm-1和1375cm-1的吸收峰分别归属于主链交联处ch2基团的面内摇摆震动和卷曲震动,分别出现了红移和蓝移现象,但是1375cm-1的谱峰吸收强度很弱。1550cm-1和1623cm-1分别归属于羧酸根coo的对称和反对称伸缩震动;3074cm-1归属于pe链交联附近ch2基团的对称伸缩振动,3116cm-1归属于pe链端附近ch2基团的反对称伸缩振动,3131cm-1归属于pe链交联附近ch2基团的反对称伸缩振动;3152cm-1的吸收峰归属于pe链端甲基团的c-h的伸缩震动。从波数为1550cm-1开始的谱峰都出现了明显的红移。
- 还没有人留言评论。精彩留言会获得点赞!