多酸基金属有机杂化材料及其制备方法和应用与流程
本发明涉及多酸基金属有机杂化材料领域。更具体地说,本发明涉及一种多酸基金属有机杂化材料及其制备方法和应用。
背景技术
多酸全称为多金属氧酸盐,是多核配合物,其中,构成多酸的的主要金属元素是钼和钨。多金属氧酸盐配合物在无机化学领域发展至今,已经有两百多年的历史了。自第一个多酸被合成发现以来,科研工作者们相继发现和合成了六种常见结构,分别为:keggin、anderson、silverton、lindqvist、dawson、waugh。
随着对于多酸研究的不断深入,其在温和条件下的良好化学选择性高催化效率逐渐显现。除此之外,多酸在应用方面还有很多突破,例如:纳米技术、医用药物、生物化学、材料化学、表面科学等。多金属氧酸盐在上述方面的进展得益于其多变的结构、高孔隙率,比表面积、孔径大小可调节、可后修饰和合成方便等。
技术实现要素:
本发明的目的是提供多酸基金属有机杂化材料及其制备方法和应用。
为了实现根据本发明的这些目的和其它优点,提供了一种含氮配体诱导的多酸基金属有机杂化材料,
通过金属盐、钼酸铵、有机配体利用蒸发回流法和/或水热合成法合成所述多酸基金属有机杂化材料;或,钼酸铵、有机配体利用蒸发回流法合成所述多酸基金属有机杂化材料;
其中,所述金属盐为铜盐或钴盐,所述有机配体为三吡啶亚甲基胺和/或1-(四氮唑-5-基)-3-(三氮唑-1-基)苯;三吡啶亚甲基胺简称为tpma,1-(四氮唑-5-基)-3-(三氮唑-1-基)苯简称为1,3-ttb。
优选的是,所述多酸基金属有机杂化材料为moo3(tpma)、2[cu(tpma)(h2o)]·(mo8o26)·4h2o、[co2(tpma)2(β-mo8o26)]或[cu2(tpma)2(1,3-ttb)(β-mo8o26)]·2h2o。
优选的是,所述moo3(tpma)的合成方法为:将0.1mmoltpma、0.1mmol钼酸铵、0.45mmol盐酸、30ml水混合放入容器中,在110℃下蒸发回流5天,冷却,过滤后得到黄褐色混浊溶液,过滤后得到淡黄色澄清溶液,将溶液置于烧杯中静置挥发后得到淡黄色长方体状晶体,即得。
优选的是,2[cu(tpma)(h2o)]·(mo8o26)·4h2o的合成方法为:将0.02mmolmoo3(tpma)、0.1mmol五水硫酸铜和10ml水放入聚四氟乙烯内衬的高压不锈钢反应釜中,用硝酸溶液调节ph至2.35,密封后将反应釜置于烘箱中,160℃反应3天,降温后得到蓝色澄清溶液,静置挥发后得带淡蓝色菱形晶体,即得。
优选的是,[co2(tpma)2(β-mo8o26)]的合成方法为:将0.1mmol硝酸钴六水合物、0.1mmoltpma、0.1mmol1,3-ttb、0.1mmol钼酸铵和10ml水放入聚四氟乙烯内衬的高压不锈钢反应釜中,用硝酸溶液调节ph至2.52,密封后将反应釜置于烘箱中,160℃反应3天,程序降温,得到酒红色块状晶体,即得。
优选的是,[cu2(tpma)2(1,3-ttb)(β-mo8o26)]·2h2o的合成方法为:将0.1mmol三水硝酸铜、0.1mmoltpma、0.1mmol1,3-ttb、0.1mmol钼酸铵和10ml水放入聚四氟乙烯内衬的高压不锈钢反应釜中,用盐酸溶液调节ph至2.35,密封后将反应釜置于烘箱中,160℃反应3天,程序降温,得到蓝色菱形晶体,即得。
本发明还提供了含氮配体诱导的多酸基金属有机杂化材料作为光催化材料降解有机染料的应用。
本发明还提供了含氮配体诱导的多酸基金属有机杂化材料作为铝土矿脱硅用抑制剂的应用。
本发明至少包括以下有益效果:利用水热合成法时向反应釜中加入金属盐、多酸、有机配体和溶剂,蒸发回流法则是向圆底烧瓶中加入金属盐、多酸、有机配体和溶剂加热回流。由于多酸阴离子具有端氧和桥氧原子配位点,所以金属阳离子可以很好的与多酸配位。再利用有机配体(有机配体中的n,o原子有孤对电子)易于金属成键,从而延伸得到丰富结构且高维度的化合物。
由于铜和钴具有灵活的配位方式和易改变的化合价,因此作为过渡金属离子。所选择的配体为三吡啶亚甲基胺(tpma)和1-(四氮唑-5-基)-3-(三氮唑-1-基)苯(1,3-ttb配体),选择三吡啶亚甲基胺(tpma)原因为:tpma存在四个潜在的配位点,有丰富的桥连模式,拥有得到多维度和高核结构的基础,并且目前与tpma相关的多酸基金属有机杂化材料的报道比较少;1,3-ttb则存在六个潜在的配位点,这使得其可以很容易的与金属配位,并得到丰富的几何构型,同时此配体含有刚性骨架,使得合成的mofs的结构相对稳定,并且在1,3-ttb配体中存在两个含氮的官能团从而使其的配位存在更多的可能性。由于上述原因所以选择tpma和1,3-ttb来尝试合成结构新颖的多酸基金属有机杂化材料。
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本发明的研究和实践而为本领域的技术人员所理解。
附图说明
图1是化合物1的红外光谱图;
图2是化合物2的红外光谱图;
图3是化合物3的红外光谱图;
图4是化合物4的红外光谱图;
图5是化合物1的循环伏安曲线图;
图6是化合物3的循环伏安曲线图;
图7是化合物4的循环伏安曲线图;
图8是化合物4对nano2的催化性质;
图9是化合物4对h2o2的催化性质;
图10是化合物1对亚甲基蓝溶液的光催化效果;
图11是化合物3对亚甲基蓝溶液的光催化效果;
图12是化合物4对亚甲基蓝溶液的光催化效果;
图13是化合物1对罗丹明b溶液的光催化效果;
图14是化合物3对罗丹明b溶液的光催化效果;
图15是化合物4对罗丹明b溶液的光催化效果;
图16是化合物1的x射线衍射图;
图17是化合物2的x射线衍射图;
图18是化合物3的x射线衍射图;
图19是化合物4的x射线衍射图;
图20是化合物1的分子结构图;
图21是化合物2的分子结构图;
图22是化合物3的分子结构图;
图23是化合物3的一维链示意图;
图24是化合物4的分子结构图;
图25是化合物2的二维结构图;
图26是化合物2的二维层简图。
具体实施方式
下面结合实施例对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
tpma合成方法为,在圆底烧瓶中加入2-甲氨基吡啶、2-氯甲基吡啶盐酸盐、碳酸钾、乙腈,在80℃条件下加热回流三天,过柱,得到黄棕色固体,即得。
实施例1
化合物1:moo3(tpma)的合成
moo3(tpma)的合成方法为:将0.1mmoltpma、0.1mmol钼酸铵、0.45mmol盐酸、30ml水混合放入容器中,在110℃下蒸发回流5天,冷却,过滤后得到黄褐色混浊溶液,过滤后得到淡黄色澄清溶液,将溶液置于烧杯中静置挥发后得到淡黄色长方体状晶体,即得。
实施例2
化合物2:2[cu(tpma)(h2o)]·(mo8o26)·4h2o
2[cu(tpma)(h2o)]·(mo8o26)·4h2o的合成方法为:将0.02mmolmoo3(tpma)、0.1mmol五水硫酸铜和10ml水放入聚四氟乙烯内衬的高压不锈钢反应釜中,用硝酸溶液调节ph至2.35,密封后将反应釜置于烘箱中,160℃反应3天,降温后得到蓝色澄清溶液,静置挥发后得带淡蓝色菱形晶体,即得。
实施例3
化合物3:[co2(tpma)2(β-mo8o26)]
[co2(tpma)2(β-mo8o26)]的合成方法为:将0.1mmol硝酸钴六水合物、0.1mmoltpma、0.1mmol1,3-ttb、0.1mmol钼酸铵和10ml水放入聚四氟乙烯内衬的高压不锈钢反应釜中,用硝酸溶液调节ph至2.52,密封后将反应釜置于烘箱中,160℃反应3天,程序降温,得到酒红色块状晶体,即得。
实施例4
化合物4:[cu2(tpma)2(1,3-ttb)(β-mo8o26)]·2h2o
[cu2(tpma)2(1,3-ttb)(β-mo8o26)]·2h2o的合成方法为:将0.1mmol三水硝酸铜、0.1mmoltpma、0.1mmol1,3-ttb、0.1mmol钼酸铵和10ml水放入聚四氟乙烯内衬的高压不锈钢反应釜中,用盐酸溶液调节ph至2.35,密封后将反应釜置于烘箱中,160℃反应3天,程序降温,得到蓝色菱形晶体,即得。
相关实验
1、晶体结构测定
如图16~19所示,化合物1~4的单晶x射线衍射数据是通过xcalibur,eos,gemini衍射仪测得,在显微镜下挑选大小合适和质量比较好的晶体进行测定。室温保持在296k,使用石墨单色化的mokα
表1化合物1–4的晶体学数据和结构参数
表2化合物1,2主要的键长
表3化合物3,4主要的键长
2、化合物1-4的晶体结构分析
化合物1:moo3(tpma)的晶体结构
如图20所示,化合物1中包含了一个tpma配体,一个moo3,结晶在斜方晶系pbca,键价计算表明所有的mo原子都是+vi氧化态。为了使结构图更加清晰,所有间隙水分子以及氢原子已被省略,之后的化合物2-4也均省略了水分子一个氢原子,不再详细说明。
在化合物1中,mo1与tpma中的三个n原子和mo8o26中的三个o原子进行6配位,形成了双三角锥形结构。结构中tpma配体中存在未配位的n,以及裸露的o原子,所以以此晶体为原料,在反应体系中继续引入第二金属或配体进行尝试。mo-o键和mo-n键的键长范围分别在
化合物2:2[cu(tpma)(h2o)]·(mo8o26)·4h2o
如图21所示,化合物2中包含了一个cu(ii)离子,一个tpma配体,一个游离的[mo8o26]4-多酸阴离子(简称mo8o26)和五个水分子。属于单斜晶系c2/2,键价计算表明,所有的mo原子都是+vi氧化态,所有的cu原子都是+ii氧化态。
如图25、26所示,在化合物2中,cu1离子与一个tpma配体和一个水进行五配位,mo8o26为游离状态,形成了五面体结构。cu-o键和cu-n键的键长范围分别在
化合物3:[co2(tpma)2(β-mo8o26)]
如图22所示,化合物3中包含了两个co(ii)离子,两个tpma配体,一个β-[mo8o26]4-多酸阴离子(简称mo8o26)。属于单斜晶系p21/c,键价计算表明,所有的mo原子都是+vi氧化态,所有的co原子都是+ii氧化态。
如图23所示,化合物3中两个co离子的配位环境都是相同且对称的。co与β-mo8o26的两个o原子(mo–o)和tpma进行六配位,形成了扭曲的八面体结构。β-[mo8o26]4-单元通过共享两个共用顶点(o)缩合形成无限的[mo8o27]n4n-链,这样的八钼链比较少见。co–o键和co–n键的键长范围分别在
化合物4:[cu2(tpma)2(1,3-ttb)(β-mo8o26)]·2h2o
如图24所示,化合物4中包含了两个cu(ii)离子,两个tpma配体,一个1,3-ttb配体,一个β-[mo8o26]4-多酸阴离子(简称mo8o26)和两个水分子。属于三斜晶系p-1,键价计算表明所有的mo原子都是+vi氧化态,所有的cu原子都是+ii氧化态。
化合物4中,三个cu(ii)离子中的cu1和cu1#的是对称的,且配位结构相同。结构中cu与两个tpma,两个1,3-ttb配体,一个β-[mo8o26]4-配位,形成了一个不规则的八面体结构。结构中三个cu(ii)离子处于同一平面切处于同一链上,通过cu(ii)离子和1,3-ttb配体配位形成一维链结构。再通过β-[mo8o26]4-中的氧与cu(ii)离子配位链接形成了二维平面结构。cu-o键和cu-n键的键长范围分别在
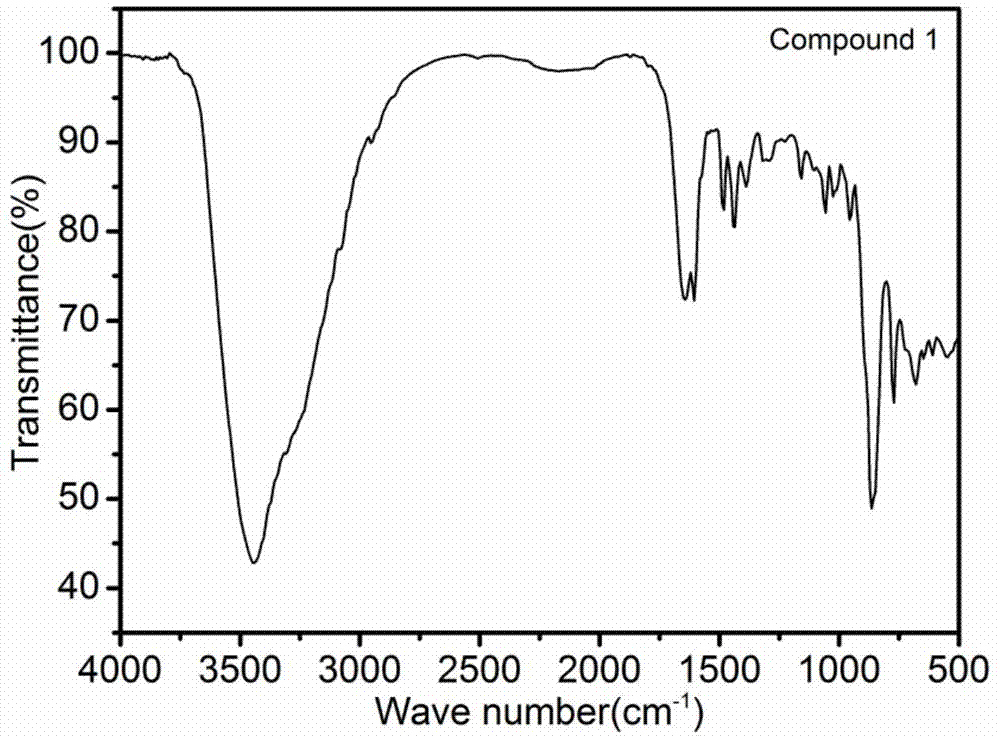
3、化合物1-4的红外光谱分析
化合物1-4的红外红外光谱图如图1~4所示。测定红外时,均用kbr与化合物以100:1的比例混合后压片通过varian640型号ft-ir光谱仪测得。通过谱图可以看出:化合物1,3343cm-1处的特征峰是羟基中的o-h伸缩振动。1159~1643cm-1处的特征峰为c-n,c=c的伸缩振动。549~1058cm-1处的特征峰可以归结于[mo8o26]4-中的mo-o和mo-o-mo的伸缩振动。化合物2,3340cm-1处的特征峰可以归结于水分子和羟基中的o-h伸缩振动。1153~1588cm-1处的特征峰可以归结于c-n,c=c的伸缩振动。694~1042cm-1处的特征峰可以归结于[mo8o26]4-中的mo-o和mo-o-mo的伸缩振动。化合物3,3431cm-1处的特征峰是羟基中的o-h伸缩振动。1152~1602cm-1处的特征峰为tpma配体中的c-n,c=c的伸缩振动。610~1056cm-1处的特征峰可以归结于[mo8o26]4-中的mo-o和mo-o-mo的伸缩振动。化合物4,3488cm-1处的特征峰可以归结于化合物中的水分子和羟基中的o-h伸缩振动。1164~1610cm-1处的特征峰为tpma配体和1,3-ttb中的c-n,c=c和n-n键的伸缩振动。528-1051cm-1处的特征峰可以归结于[mo8o26]4-中的mo-o和mo-o-mo的伸缩振动。
4、化合物1,化合物3和化合物4的电化学性质分析
由于化合物2在电解液中不稳定,所以重点探究了化合物1,化合物3和化合物4的电化学性质,如图5~7所示。首先分别将化合物1-4称取少量后与石墨粉混合后在玛瑙研钵中研磨至混合均匀,用硅油将其搅拌至粘稠后,装入玻璃管中,完成后插入铜丝待测。检测所用的电化学工作站为chi660e电化学工作站,所用的参比电极为ag/agcl电极,对电极为铂电极,电解液为硫酸与硫酸钠混合溶液,最后测得的循环伏安曲线如图所示。在0-500mv的电位范围内,化合物1在200mv处存在一对氧化还原峰(e1/2=(epa+epc)/2)对应的是mo8o26的还原过程。化合物3在200mv处和350mv处存在两对氧化还原峰(e1/2=(epa+epc)/2)对应的是mo8o26的还原过程。化合物4在75mv处存在一对氧化还原峰(e1/2=(epa+epc)/2)对应的是cu的电子氧化还原过程,在200mv处存在一对氧化还原峰(e1/2=(epa+epc)/2)对应的是mo8o26的还原过程。
在基于上述基础上,进一步研究了化合物4对nano2和h2o2的催化性质的研究。结果如图8~9所示。根据图氧化还原峰的变化情况进行分析,可以看出其氧化还原峰随着nano2和h2o2的增加不断上移判断其具有催化nano2和h2o2的性质。
5、化合物1~4的光催化性能
对于光催化性能的研究,选择了亚甲基蓝和罗丹明b作为污染物来研究不同晶体对染料的降解性质。实验步骤为,称取少量晶体于烧杯中,加入亚甲基蓝(6mg/l-1)或罗丹明b(10mg/l-1),再向另一个烧杯中加入亚甲基蓝(6mg/l-1)或罗丹明b(10mg/l-1)作为空白对照。将烧杯置于紫外灯照射下的磁力搅拌器上,每隔半小时取出一部分反应溶液进行紫外测定。
如图10~12所示,经紫外测定后分析图可以得到化合物1~4对亚甲基蓝都具有催化降解作用,四个化合物中,化合物1的催化效率最高,特别是在前半个小时,化合物3次之,化合物2和化合物4的催化效率均很低。如图13~15所示,化合物1、3、4均对罗丹明b有较高的催化效率,其中化合物4对罗丹明b的催化降解效果很显著,化合物3次之,化合物1比较差。
6、化合物1作为铝土矿脱硅用抑制剂的应用
捕收剂采用油酸钠。
抑制剂1号采用水玻璃。
抑制剂2号为水玻璃和化合物1按质量比100:3混合后3000r/min搅拌15min制成,搅拌温度控制在20℃。
铝土矿原矿中al2o3含量为40.13%,sio2含量为8.66%,铝土矿原矿中的脉石矿物为石英、水云母、绿泥石、方解石等。将铝土矿原矿用球磨机进行磨矿,磨矿细度为-0.074mm占70%,矿浆浓度为34%,进行一粗两扫流程,浮选药剂用量:一次粗选加入调节剂为na2co31500g/t,抑制剂为600g/t,捕收剂150g/t,两次扫选均为加入捕收剂8g/t,将粗精矿和扫选得到的中矿合并两次精选流程,结果见表1。
表1
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
- 还没有人留言评论。精彩留言会获得点赞!