一种二吡啶基吡咯-钌(I)配合物及其制备方法和作为电化学还原催化剂的应用与流程
本发明涉及一种[ru2(co)4(pdp)2]配合物,还涉及通过十二羰基三钌氧化或二氯·二羰基合钌(ii)多聚物还原与2,5-二吡啶基吡咯配体(hpdp)反应制备[ru2(co)4(pdp)2]配合物的方法,还涉及[ru2(co)4(pdp)2]配合物作为co2电化学催化还原的均相催化剂的应用,属于均相催化技术领域。
背景技术
[ru2(co)4]2+配合物是底物活化和均相催化中的中间体,比如co2催化还原的活性中间体。chardon-noblat等人(chardon-noblats,deronziera,ziesselr,etal.selectivesynthesisandelectrochemicalbehavioroftrans(cl)-andcis(cl)-[ru(bpy)(co)2cl2]complexes(bpy=2,2‘-bipyridine).comparativestudiesoftheirelectrocatalyticactivitytowardthereductionofcarbondioxide[j].inorganicchemistry,1997,36(23):5384-5389.)发现cis(cl)-[ru(bpy)(co)2cl2]和cis(co)-[ru(bpy)(co)2(c(o)ome)cl]都是二氧化碳电还原的前催化剂。他们提出,cl-的离去导致[ru2(co)4]2+的形成,其催化co2还原成co和痕量的甲酸盐。ishida报道了在dma/h2o混合物中trans(cl)-[ru(bpy)(co)2cl2]与[ru(bpy)3]2+的光敏反应(kuramochiy,itabashij,fukayak,etal.unexpectedeffectofcatalystconcentrationonphotochemicalco2reductionbytrans(cl)–ru(bpy)(co)2cl2:newmechanisticinsightintotheco/hcoo-selectivity[j].chemicalscience,2015,6(5):3063-3074.)。co和甲酸盐产生的比例取决于催化剂浓度和光照强度。作者通过引入[ru2(co)4]2+的形成来解释这种选择性,[ru2(co)4]2+负责产生甲酸,同时提出单核催化剂产生co。当催化剂浓度增大时,催化剂的一电子还原物质不能得到更多的电子,倾向生成[ru2(co)4]2+,co/hcoo-值减小;当光照强度减小时,co/hcoo-值也减小,显示出[ru2(co)4]2+作为催化循环中的关键活性中间体。为了验证这一假设,作者合成了反式trans(cl)-[ru(mesbpy)(co)2cl2],其中[ru2(co)4]2+的形成受到bpy配体上的甲基基团的阻碍。在这种情况下,光催化二氧化碳还原选择性地生成co,只有痕量的甲酸盐。kubia在2015年在研究该催化剂的电化学二氧化碳还原时证实了[ru2(co)4]2+形成的抑制作用(machancw,sampsonmd,kubiakcp.amolecularrutheniumelectrocatalystforthereductionofcarbondioxidetocoandformate[j].journaloftheamericanchemicalsociety,2015,137(26):8564-8571.)。co是苯酚作为质子源的控制电压电解(cpe)中的主要产物,尽管这种情况下产物选择性决定于采用的电压。
总之,钌-多吡啶体系这类化合物具有合成可调控性,稳定性和对co2还原催化作用的高活性。但是,尽管多吡啶可能会被合成修饰以提供具有各种电子和空间效应的各种配体,但是在研究最多的配体的结构中看到如此小的变化是令人惊讶的,例如联吡啶、菲咯啉和三联吡啶。取代基通常限于甲基,叔丁基和羧基,这是由于这样的事实,即只有少数这样的衍生物在市场上可获取,并且在开发用于制备联吡啶、菲咯啉和三联吡啶配体的原始衍生物的合成策略方面的努力太少。目前以它们为基础的双核ru(i)-ru(i)配合物主要通过电化学手段合成,或者为光/电催化过程中形成的中间体,制备方法较复杂。
技术实现要素:
针对现有技术中的钌-多吡啶体系存在的上述缺陷,本发明的第一个目的是在于提供一种稳定性好,co2还原催化作用活性高的[ru2(co)4(pdp)2]配合物。
针对现有[ru2(co)4]2+配合物的合成[ru2(co)4]2+方法存在能耗高,操作复杂等缺点,本发明的第二个目的是在于提供一种合成[ru2(co)4(pdp)2]的方法,该方法丰富了[ru2(co)4]2+配合物合成,步骤简单,易于操作,有利于扩大生产。
本发明的第三个目的是在于[ru2(co)4(pdp)2]配合物作为电化学还原co2催化剂的应用,其表现出较高的催化活性。
为了实现上述技术目的,本发明提供了一种[ru2(co)4(pdp)2]配合物,其具有式1结构:
其中,r1和r2独立选自氢或卤素。
优选的[ru2(co)4(pdp)2]配合物为
本发明还提供了[ru2(co)4(pdp)2]配合物的合成方法,其包括以下两种合成方法:
方案一:2,5-二吡啶基吡咯和十二羰基三钌在甲苯中回流反应,即得;
方案二:2,5-二吡啶基吡咯与二氯·二羰基合钌(ii)多聚物及缚酸剂在甲醇中回流反应,即得。
优选的方案,方案一中,2,5-二吡啶基吡咯配体的摩尔量为十二羰基三钌摩尔量的3~4倍。
优选的方案,方案一中,回流反应温度为110~120℃,时间为8~12h。
优选的方案,方案二中,2,5-二吡啶基吡咯配体的摩尔量为二氯·二羰基合钌(ii)多聚物摩尔量的1~2倍。
优选的方案,方案二中,缚酸剂的摩尔量为2,5-二吡啶基吡咯摩尔量的3~4倍。缚酸剂为三乙胺。
优选的方案,方案二中,回流反应的温度为70~90℃,时间为8~12h。
本发明还提供了[ru2(co)4(pdp)2]配合物的应用,其作为均相催化剂应用于co2电化学催化还原。
本发明的[ru2(co)4(pdp)2]配合物主要配体为2,5-二吡啶基吡咯,其是一种多吡啶多齿配体,具有丰富的配位模式,能够稳定金属中心,且通过电子效应,提高配合物的催化活性。
本发明的[ru2(co)4(pdp)2]双核配合物作为二氧化碳还原催化剂与其他多吡啶钌羰基配合物相比,含有ru-ru金属键,ru处于+1低价态,与后者通过还原催化循环产生的[ru2(co)4]2+双核催化活性中间体结构类似,理论上还原产物hcooh选择性将更高,催化性能更好。
本发明[ru2(co)4(pdp)2]配合物的合成方法,提出了两种合成思路。第一种思路为:以十二羰基三钌为前体,与2,5-二吡啶基吡咯配体(hpdp)在甲苯中回流,通过十二羰基三钌氧化得到[ru2(co)4(pdp)2]配合物;该方法巧妙地以2,5-二吡啶基吡咯配体(hpdp)上的酰胺质子作为氧化剂,将十二羰基三钌中的0价钌氧化为+1价钌。第二种思路为:通过与2,5-二吡啶基吡咯配体(hpdp)在缚酸剂(三乙胺)存在下于甲醇中反应还原二氯·二羰基合钌(ii)多聚物制备,得到配合物[ru2(co)4(pdp)2];其中甲醇具有两个重要的作用,一方面作为溶剂,另一方面作为还原剂,将前体+2价钌还原为+1价钌。
十二羰基三钌:
2,5-二吡啶基吡咯配体:
[ru2(co)4(pdp)2]配合物:
其中,上述结构式中r1为h,r2为h;或者,r1为cl,r2为h;或者r1为h,r2为br。
相对现有技术,本发明的技术方案带来的有益技术效果:
1)本发明的[ru2(co)4(pdp)2]配合物采用2,5-二吡啶基吡咯多齿配体,具有丰富的配位模式,能够稳定金属中心,且通过电子效应,提高配合物的催化活性,且含有ru-ru金属键,ru处于+1低价态,相对现有的类似配合物具有更好的稳定性和更高的催化活性。
2)本发明提出了两种全新的合成[ru2(co)4(pdp)2]配合物的方法,由2,5-二吡啶基吡咯配体(hpdp)与十二羰基三钌经氧化,或者由2,5-二吡啶基吡咯配体(hpdp)与二氯·二羰基合钌(ii)多聚物经还原得到[ru2(co)4]2+配合物。具有步骤简单,反应条件温和,易于操作等优点,有利于大规模生产应用。
3)本发明的[ru2(co)4(pdp)2]配合物作为均相催化剂,相比于其他单核ru(ii)配合物,含有ru(i)-ru(i)金属键,稳定性较好,表现出较高的co2电化学催化还原性能,具有广泛应用前景。
附图说明
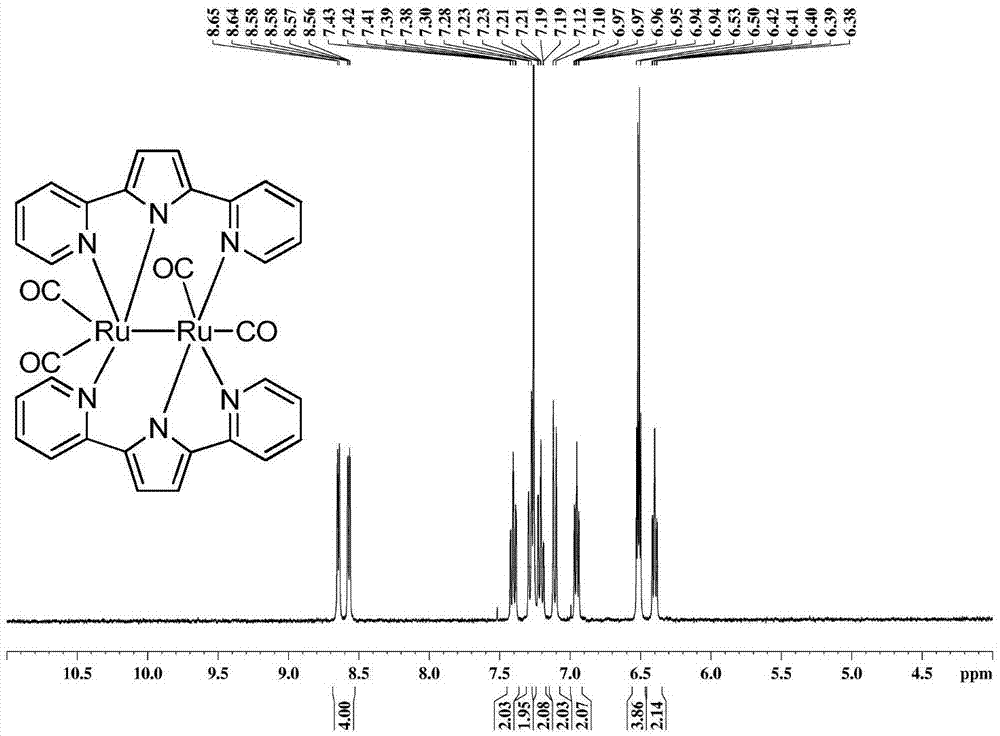
【图1】为实施例1中配合物1[ru2(co)4(l1)2]的1hnmr图谱;
【图2】为实施例1中配合物1[ru2(co)4(l1)2]的ir图谱;
【图3】为实施例1中配合物1[ru2(co)4(l1)2]的单晶结构椭球图;
【图4】为实施例3中配合物2[ru2(co)4(l2)2]的1hnmr图谱;
【图5】为实施例3中配合物2[ru2(co)4(l2)2]的ir图谱;
【图6】为实施例3中配合物2[ru2(co)4(l2)2]的单晶结构椭球图;
【图7】为实施例4中配合物3[ru2(co)4(l3)2]的1hnmr图谱;
【图8】为实施例4中配合物3[ru2(co)4(l3)2]的ir图谱;
【图9】为实施例4中配合物3[ru2(co)4(l3)2]的单晶结构椭球图;
【图10】为实施例1~4中配合物1~3在二氯甲烷溶液中的紫外可见光谱图;
【图11】为实施例5中配合物1在ar及co2氛围下的电化学循环伏安曲线图;
具体实施方式
以下实施例旨在进一步说明本发明内容,而不是限制本发明权利要求的保护范围。
以下实施例中的化合物,如没有特殊说明,原料均为市售原料。
实施例1~4与对比实施例1~3主要说明[ru2(co)4(pdp)2]配合物的合成。
实施例5主要说明[ru2(co)4(pdp)2]作为均相催化剂催化co2电化学还原的性质。
以下实施例中涉及的底物原料,以及溶剂等均为市售商业产品(分析纯试剂),所用试剂均经过纯化、干燥及除氧预处理,涉及的合成及处理过程使用标准无水无氧技术。其中二氯·二羰基合钌(ii)多聚物(andersonpa,deacongb,haarmannkh,etal.designedsynthesisofmononucleartris(heteroleptic)rutheniumcomplexescontainingbidentatepolypyridylligands[j].inorganicchemistry,1995,34(24):6145-6157.)及2,5-二吡啶基吡咯(hpdp)(imlergh,luz,kistlerka,etal.complexesof2,5-bis(α-pyridyl)pyrrolatewithpd(ii)andpt(ii):amonoanioniciso-π-electronligandanalogofterpyridine[j].inorganicchemistry,2012,51(19):10122-10128.)按已有报道方法合成。
1hnmr(400mhz),以cdcl3为溶剂,以tms为内标。
多重性定义如下:s(单峰);d(二重峰);t(三重峰);q(四重峰)和m(多重峰)。偶合常数j(赫兹)。
ir,kbr压片。吸收强度定义如下:s(强吸收);m(中等程度吸收);w(弱吸收)。
合成方法一:
在氮气氛围下,将配体2,5-二吡啶基吡咯(hpdp)(2.4mmol)和ru3(co)12(0.6mmol)置于100ml两口圆底烧瓶中,加入已充分除氧的甲苯(40ml),搅拌下加热至回流,ru3(co)12逐渐溶解,得到深红色澄清溶液。持续搅拌回流过夜,有黄色不溶物生成,反应完成。待反应体系冷却至室温后,过滤移去溶液,收集黄色沉淀,分别用冷的甲苯(5ml*3)及乙醚(5ml*3)洗涤,真空下干燥,得到产物。
合成方法二:
在氮气氛围下,将配体2,5-二吡啶基吡咯(hpdp)(1mmol)与多聚物[rucl2(co)2]n(1mmol)置于50ml两口圆底烧瓶中,加入已充分除氧的甲醇(30ml),干燥过的三乙胺(3.6mmol)作为去质子碱。反应混合液于搅拌下加热至回流,固体逐渐溶解得到棕红色澄清溶液。持续搅拌回流过夜,有黄色不溶物生成,反应完成。待反应体系冷却至室温后,过滤后得到黄色沉淀,分别用冷的甲醇(5ml*3)及乙醚(5ml*3)洗涤,抽干,得到产物。
以下实施例1~4按照上述反应条件进行反应:
实施例1
按方法一合成:
原料:2,5-二吡啶基吡咯(hl1),十二羰基三钌;
目标产物:
1hnmr(400mhz,cdcl3):δ8.65(d,j=5.04hz,2h),8.58(dd,j=5.84,1.08hz,2h),7.43(td,j=8.24,1.52hz,2h),7.30(d,j=7.68hz,2h),7.23(td,j=7.04,1.60hz,2h),7.12(d,j=8.16hz,2h),6.97(td,j=5.48,1.12hz,2h),6.53(q,j=3.92hz,4h),6.42(td,j=7.20,1.52hz,2h);
ir(kbr,cm-1):2010(s),1967(s),1925(s),1600(m),1552(w),1505(m),1456(w),1429(m),1339(m),1157(w),963(w),780(w),746(m),650(w),586(w),538(w)。
实施例2
按方法二合成:
原料:2,5-二吡啶基吡咯(hl1),[ru(co)2cl2]n,三乙胺;
目标产物:
表征数据同方法一中实施例1。
实施例3
按方法二合成:
原料:3,4-二氯-2,5-二吡啶基吡咯(hl2),[ru(co)2cl2]n,三乙胺;
目标产物:
1hnmr(400mhz,cdcl3):δ8.77(d,j=4.84hz,2h),8.65(dd,j=5.84,0.96hz,2h),7.87(d,j=8.24hz,2h),7.81(d,j=8.24hz,2h),7.56(td,j=7.24,1.36hz,2h),7.45(td,j=8.68,1.64hz,2h),7.09(td,j=5.52,1.04hz,2h),6.67(td,j=8.76,1.52hz,2h);
ir(kbr,cm-1):2016(s),1970(s),1936(s),1915(s),1597(s),1556(w),1492(m),1432(m),1336(s),1159(w),972(w),776(w),744(w),647(w),571(w),533(w)。
实施例4
按方法二合成:
原料:2,5-二(5,5’-二溴)吡啶基吡咯(hl3),[ru(co)2cl2]n,三乙胺;
目标产物:
收率:25%;
1hnmr(400mhz,cdcl3):δ8.74(d,j=1.92hz,2h),8.60(d,j=2.12hz,2h),7.61(dd,j=8.64,2.04hz,2h),7.33(dd,j=8.84,2.20hz,2h),7.16(d,j=8.88hz,2h),7.04(d,j=8.64hz,2h),6.51(d,j=3.76hz,2h),6.48(d,j=3.88hz,2h);
ir(kbr,cm-1):2021(s),1977(s),1927(s),1583(m),1504(s),1444(m),1369(s),1327(m),1230(w),1140(w),918(w),827(w),760(w),729(w),650(w),588(w),534(w)。
实施例5
取3.7mg[ru2(co)4(l1)2]溶于5ml乙腈溶液中(含0.1m四丁基六氟磷酸铵作为支持电解质),配置成1mm配合物溶液,进行循环伏安测试。测试采用三电极体系,玻碳电极为工作电极、铂丝电极为对电极、ag/agno3电极作为参比电极。循环伏安测试分别于惰性气氛(ar)及co2氛围下进行。
为了说明取代基团为其他较大基团或不同位置取代时,难以得到配合物的情况,采用其他较大基团或不同位置取代的2,5-二吡啶基吡咯配体以方法二同样条件来合成目标配合物[ru2(co)4(pdp)2],见对比实施例1~3。
对比实施例1
按方法二合成:
原料:2,5-二(6,6’-二溴)吡啶基吡咯(hl4),[ru(co)2cl2]n,三乙胺;
2,5-二(6,6’-二溴)吡啶基吡咯(hl4):
未得到目标配合物。
对比实施例2
按方法二合成:
原料:2,5-二(5,5’-二苯基)吡啶基吡咯(hl5),[ru(co)2cl2]n,三乙胺;
2,5-二(5,5’-二苯基)吡啶基吡咯(hl5):
未得到目标配合物。
对比实施例3
按方法二合成:
原料:2,5-二(5,5’-二噻吩)吡啶基吡咯(hl6),[ru(co)2cl2]n,三乙胺;
2,5-二(5,5’-二噻吩)吡啶基吡咯(hl6):
未得到目标配合物。
图1~图9分别为配合物1~3的核磁、红外、单晶结构;图10为配合物1~3的紫外可见光谱图;图11展示了配合物1在ar及co2氛围下的循环伏安曲线图,从中可以看出,epa=-2.50v处的还原峰电流在co2氛围下相比ar氛围有大幅度增大,为二氧化碳还原过程,说明配合物1对co2电化学催化还原具有较高活性。
表1实施例1~4中配合物1~3的晶体学数据表
表2实施例1~4中配合物1~3的部分键长键角数据
以上显示和描述了本发明制备[ru2(co)4]2+配合物的主要方法特征和优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理和方法过程,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种含有氟氯吡啶酯和氯吡嘧磺隆的除草剂组合物的制作方法
- (-)-(2r,3s)-2-氨基-3-羟基-3-吡啶-4-基-1-吡咯烷-1-基-丙-1-酮(l)-(+)酒石酸盐 ...的制作方法
- 吡咯并吡啶或吡唑并吡啶衍生物的制作方法
- 吡啶胺基嘧啶衍生物、其制备方法及应用
- 作为lrrk2抑制剂的芳基吡咯并吡啶衍生的化合物的制作方法
- 四氢吡啶并[4,3-d]嘧啶类Hsp90抑制剂及其医药用图
- 具有催化二氢嘧啶酮类化合物的吡啶乙烯配位聚合物的制作方法
- 用于Knoevenagel缩合反应的层状石墨相氮化碳/负载型催化剂及其制备方法
- 1h-吡咯并[2,3-b]吡啶-3-羧酸甲酯的合成工艺的制作方法
- 一种新的吡啶酮类生物碱及其制备方法