一种混合C4生产二异丁烯的方法与流程
一种混合c4生产二异丁烯的方法,属于c4聚合技术领域。
背景技术:
二聚异丁烯(二异丁烯)主要成份为2,4,4-三甲基-1-戊烯和2,4,4-三甲基-2-戊烯,是无色透明液体,易挥发。二异丁烯做为宝贵的化工原料越来越受到人们的重视。可用于生产辛基酚、异壬醇及辛基二苯基胺等多种精细化工产品。二异丁烯和苯酚反应生成辛基酚。后者是一种重要的精细化工产品,是合成表面活性剂抗氧剂、稳定剂、树脂等的基本原料。
石油加工过程中产生大量混合c4,其中含有1%~99.99%的异丁烯,原料中的异丁烯聚合生成聚合产品,主要成分为二异丁烯,同时含有少量其他c8烯烃,将聚合产品分离得到二异丁烯。有关混合c4中异丁烯聚合利用的专利和文章很多。us4100220在原料中配入适量叔丁醇来提高聚合反应选择性,并设反应器外循环以取走反应热,由于循环物料中含有烯烃,所以产品中高聚物含量较高。cn1087616a公开了混合碳四的数级固定床串联聚合工艺,原料依次经过两级或两级以上串联反应器,反应催化剂为硅酸铝小球,通过反应温度来控制聚合反应进行,由于没有添加聚合反应抑制剂,同样使得反应产品中三聚物含量较高,二聚物及c8含量只能达到65%以上。us4215011采用催化蒸馏工艺进行混合c4中异丁烯地选择性聚合,催化蒸馏工艺可以解决很多固定床中不容易解决的问题。蒸馏塔内各点的温度取决于设定压力下物料的沸点,不会出现反应床层温升过高的问题。回流液的冲刷作用较强,避免了聚合物粘附、聚集在催化剂表面。分离作用使二聚产物很快离开催化剂表面,减少进一步聚合的机会,从而提高二聚反应的选择性。该专利没有使用水或醇类来提高聚合反应选择性,反而认为原料中含有的少量水会对异丁烯的选择性二聚不利,这一点与其他文献介绍的工业化实施例相左。us4375576公开了一种碳四中的异丁烯液相反应,催化剂为离子交换树脂,采用mtbe作抑制剂,抑制高聚物、共聚物等副产物的生成。us2008242909公开了一种在有碳五和含氧化合物存在的情况下异丁烯选择性二聚生产高辛烷值组分的方法,含氧化合物为醇类或醚类,第二步反应形式为催化蒸馏。cn1210379公开了利用混合c4为原料在催化剂及有抑制剂的存在下发生聚合反应,抑制剂是水、叔丁醇或其混合物,聚合反应后经水洗、回收抑制剂得到的聚合产品进行加氢反应得到烷基化产品。聚合反应经过固定床预聚反应和催化蒸馏反应。该专利使c4中异丁烯的转化率达到90%以上,加氢产品中c8总含量≥92%,辛烷值≥95。上述技术要么没有添加抑制剂,要么添加的抑制剂为水、醇类或醚类,聚合产品中二异丁烯含量不高,或者抑制剂不易回收。
技术实现要素:
本发明要解决的技术问题是:克服现有技术的不足,提供一种产品选择性高、抑制剂方便回收的混合c4生产二异丁烯的方法。
本发明解决其技术问题所采用的技术方案是:该混合c4生产二异丁烯的方法,其特征在于:混合c4在催化剂及有抑制剂的存在下发生聚合反应,聚合反应的温度为20℃~140℃,反应压力为0.2mpa~4.0mpa,所述的混合c4含有质量分数1%~99.99%的异丁烯,所述催化剂为大孔磺酸阳离子交换树脂,大孔磺酸阳离子交换树脂以h+计交换容量为4.0mmol/g~5.5mmol/g,使用空速为0.2h-1~8h-1;所述抑制剂为丙酮和丁酮的混合物,抑制剂与混合c4中的异丁烯的摩尔比为:0.003~1.5:1;聚合反应后的聚合产品经过分离得到二异丁烯,聚合反应后通过蒸馏回收抑制剂。
本发明所要解决的技术问题是利用混合c4生产二异丁烯的方法。在酸性催化剂条件下,混合碳四中的异丁烯二聚反应比较剧烈,同时会有三聚甚至四聚反应发生,为了抑制三聚以上反应,通常加入一定的抑制剂提高二聚反应选择性。但二聚反应选择性提高的同时,转化率必然下降很多。发明人发现,在反应体系中加入丙酮和丁酮的混合物,并与特定的树脂催化剂配合,能同时达到较高的反应转化率和二异丁烯反应选择性。本发明将混合c4中的异丁烯、少量丁烯-1在高效催化剂和抑制剂作用下高转化率、高选择性二聚,大大提高二异丁烯的收率,聚合反应后经过分离得到二异丁烯,抑制剂不参与反应,容易回收,循环利用。
聚合反应后通过蒸馏回收抑制剂。丙酮或丁酮在反应过程中不参与反应,将其回收后可循环利用。其回收方式与其他抑制剂相比更加简便,不必使用水洗的方式,既产生工业废水又存在设备腐蚀问题。在聚合产物分离过程中采用精馏或抽侧线的方式分离回收即可。流程简化,节省费用。
优选的,所述的抑制剂与混合c4中的异丁烯的摩尔比为0.01~1.0:1。优选的抑制剂的用量能够充分的保证二异丁烯反应选择性。
优选的,所述的聚合反应的温度为30℃~90℃,反应压力为0.5mpa~1.6mpa。
优选的,所述的丙酮和丁酮质量比为0.1~1:1,更加优选的,丙酮和丁酮质量比为0.2~0.4:1,所述大孔磺酸阳离子交换树脂以h+计交换容量为4.5mmol/g-5.0mmol/g。催化剂可以是市售的任何型号的酸性阳离子交换树脂催化剂,更加优选的,催化剂的制备方法包括以下步骤:
(1)按照重量份数计,在溶有0.1~10份羟丙基甲基纤维素、0.2~10聚丙烯酰胺、2~10份仲烷基磺酸钠的水中,加入15~85份致孔剂、0.2-10份过氧化苯甲酰、0.2-10份过氧化新癸酸叔丁酯、60~95份苯乙烯单体、5~40份多乙烯基单体的混合物,在反应温度40~90℃下发生聚合反应,得到产物共聚物白球,烘干后将致孔剂抽提干净;
(2)在温度为10~85℃和搅拌条件下,向共聚物白球中加入发烟硫酸,所加发烟硫酸重量为共聚物白球重量的4~10倍,然后升温至85~140℃,反应5~11小时,得到具有高温热稳定性的阳离子交换树脂。
步骤(1)中的水的用量优选为300~500份。步骤(1)中的致孔剂的用量优选为20~50份。它能与聚合单体互溶而不溶于水相,因此,聚合水相中几乎没有残留的致孔剂,省去了从聚合废水中回收致孔剂的步骤。致孔剂优选自c12-c24饱和烷烃中的一种或两种以上任意比例混合的混合物。步骤(1)中的多乙烯基单体优选自二乙烯基苯或二乙烯基甲苯。
优选配比的抑制剂能更有效抑制三聚以上高聚反应的进行,提高聚合反应的选择性,提高二异丁烯的收率。配合优选的催化剂同时实现更好的反应转化率。
本发明所述聚合反应最好经过两级或两级以上,以提高c4中烯烃的转化率及选择性。该聚合反应可以采用如下两种方式。
优选的,所述的聚合反应采用固定床反应器和催化蒸馏塔的组合聚合系统,其中固定床反应器设一台或几台,先经过固定床预聚反应,再进行催化蒸馏反应。预聚反应能够提高聚合反应的选择性,在固定床反应器中进行,减少三聚以上高聚物的生成。固定床聚合反应与催化蒸馏反应可在相同温度下操作,也可在不同温度下操作。
优选的,当c4中异丁烯的浓度在4%以上时,将固定床预聚反应的聚合反应产物冷却后部分循环回固定床聚合反应器进料口以稀释进料中异丁烯浓度并取走反应热,以重量份计聚合反应产物的循环量为混合c4进料的0.1~15倍,反应后剩余的c4在催化蒸馏塔塔顶采出;回收抑制剂,聚合产品送入二异丁烯精制塔,分离提纯二异丁烯。
当混合c4中异丁烯的浓度不超过4%时,可以不设循环进料。循环回固定床聚合反应器的物料与新鲜进料的比例与原料中异丁烯的浓度有关,异丁烯的浓度越高,需要的循环比例越大。
优选的,抑制剂在固定床预聚反应和催化反应两个阶段分别加入,其中固定床预聚反应阶段的加入量为抑制剂总加入量的60%以上。
优选的,混合c4在装有催化剂的至少两级串联固定床反应器中发生聚合反应,当混合c4中异丁烯的浓度在4%以上时从一级固定床聚合反应器出来的聚合反应产物冷却后一部分循环回一级固定床聚合反应器以稀释进料中异丁烯浓度并取走反应热,以重量份计聚合反应产物的循环量为混合c4进料的0.1~15倍,剩余部分进入二级固定床聚合反应器继续反应,产物送脱c4塔。
聚合过程中有丙酮或丁酮或其混合物作为聚合反应抑制剂存在,抑制剂的加入方式可以是一次性在第一级反应时加入,也可以是在两级反应中都加入。从一级固定床聚合反应器出来的主要组成为剩余c4、二异丁烯、异丁烯与正丁烯的共聚物、少量三聚以上高聚物的聚合产品冷却后一部分循环回一级固定床聚合反应器以稀释进料中异丁烯浓度并取走反应热,一部分进入二级固定床聚合反应器继续反应,产物送脱c4塔(可用催化蒸馏塔作为分离塔,不装催化剂进行分离)。异丁烯的浓度不超过4%时,可以不设剩余c4循环进料。循环回固定床聚合反应器的剩余c4与新鲜进料的比例与原料中异丁烯的浓度有关,异丁烯的浓度越高,需要的循环比例越大。脱c4塔的操作温度和压力同聚合反应条件。
优选的,所述的脱c4塔的聚合反应的温度为20~140℃,反应压力为0.2~4.0mpa;剩余c4在脱c4塔塔顶逸出,塔釜聚合产品回收抑制剂,聚合产品入二异丁烯精制塔,分离提纯二异丁烯。塔釜聚合产品送抑制剂回收塔精馏回收抑制剂,或从催化蒸馏塔中部抽侧线回收抑制剂,聚合产品主要成分为c8烯烃、少量c12烯烃,送入二异丁烯精制塔,分离提纯二异丁烯。
优选的,所述的聚合产品中c8总含量≥95%,c12含量≤5%,c16含量为0。
所述混合c4可以是异丁烯含量低的混合c4,也可以是高纯度异丁烯碳四,如通过mtbe分解反应得到的高纯度异丁烯,其中的异丁烯含量最高可达99%以上,即所述混合c4可以含有1~99.99%的异丁烯。
与现有技术相比,本发明的所具有的有益效果是:本发明以石油加工、化工生产过程中产生的混合c4为原料,将其中的异丁烯、少量丁烯-1在高效催化剂和抑制剂共同作用下高转化率、高选择性二聚,聚合反应后经过分离得到二异丁烯。本发明的工艺能够使c4中异丁烯的转化率达到93%以上,二异丁烯含量≥80%,分离后的二异丁烯纯度≥98%。同时抑制剂的回收方式简便,不产生工业废水和设备腐蚀,能耗低。流程简化,节省费用。本发明适用于石油烃混合物中的烯烃聚合,也适用于化工生产过程中产生的高纯度烯烃聚合,特别适用于混合c4中的烯烃聚合生产二异丁烯。
附图说明
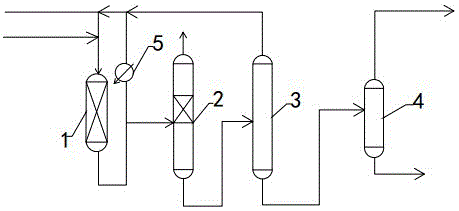
图1为本发明催化蒸馏法工艺流程图。
图2为本发明串联固定床法工艺流程图。
其中,1、固定床反应器101、一级固定床反应器、102二级固定床反应器2、催化蒸馏塔3、抑制剂回收塔4、二异丁烯精制塔5、冷凝器。
具体实施方式
下面结合具体实施例对本发明做进一步说明。
以下结合工艺流程图说明本发明:
参照附图1,催化蒸馏法工艺:混合c4和聚合反应抑制剂分别经管线和管线送入固定床反应器1,预聚合后的混合物料分成两部分,一部分经管线送入催化蒸馏塔2,一部分经冷凝器5冷凝由管线循环回固定床聚合反应器,以稀释进料烯烃浓度,降低反应温升。固定床反应器1和催化蒸馏塔2里面装填相同的聚合反应催化剂。混合c4在催化蒸馏塔2里进一步聚合,聚合后剩余c4经管线出装置,含抑制剂的聚合产品经管线出塔釜并送入抑制剂回收塔3。该塔塔顶蒸出的抑制剂由管线循环回固定床聚合反应器利用。塔釜液为聚合产品经管线送入二异丁烯精制塔4。塔釜重组分由管线连续采出,塔顶二异丁烯经管线出装置。
催化蒸馏塔的反应段可以使用任何已知形式的催化蒸馏设备。
参照附图2,串联固定床法间接烷基化工艺:混合c4和聚合反应抑制剂分别经管线送入一级固定床反应器101,初步聚合后送入冷凝器5冷凝后分成两部分,一部分由管线循环回固定床聚合反应器,以稀释进料烯烃浓度,降低反应温升。一部分经管线进入二级固定床反应器102,同时将适量的聚合反应抑制剂配入管线。混合c4在二级固定床反应器102进一步聚合,聚合后混合物料经管线送入催化蒸馏塔2,剩余c4从精馏塔塔顶经管线出装置,从精馏塔中部抽出一股侧线物料,其中主要成分为抑制剂,由管线循环回固定床聚合反应器利用。塔釜液为聚合产品经管线送入二异丁烯精制塔4。塔釜重组分由管线连续采出,塔顶二异丁烯经管线出装置。
实施例1
采用如图1的催化蒸馏法工艺;
混合c4中含异丁烯18.5%,含正丁烯16.3%,进料量:300g/h;
固定床聚合反应器催化剂装量:500ml,催化剂为大孔磺酸阳离子交换树脂,交换容量为4.5mmol〔h+〕/g;聚合反应催化剂制备方法包括以下步骤:
(1)向500g水中加入0.2g羟丙基甲基纤维素、0.3g聚丙烯酰胺、7g仲烷基磺酸钠,加热搅拌,使其完全溶解,形成均匀的水溶液。再加入由90g苯乙烯、35g二乙烯基苯、0.9克过氧化苯甲酰、24g正十六烷、6g过氧化新癸酸叔丁酯组成的有机相混合物,调节适当的搅拌转速,升温至65℃,恒温反应2小时,再升温至80℃,恒温反应4小时。将反应产物过滤,再置于3倍体积的自来水中搅拌5分钟,洗涤残余的分散剂,过滤,并在室温下晾干(含水量≤3%),得到干燥的共聚物白球,将共聚物白球中的致孔剂抽提干净;
(2)在温度为80℃和搅拌条件下,向500g共聚物白球中加入3500g发烟硫酸,搅拌升温至80℃,恒温反应3小时,再缓慢升温至95℃,恒温反应2小时,再缓慢升温至120℃,恒温反应4小时,将产物离心过滤得到阳离子交换树脂。
催化蒸馏塔催化剂装量:200ml;
原料中配入聚合反应抑制剂,抑制剂与新鲜进料中异丁烯的摩尔比为0.01∶1;
聚合产品送二异丁烯精制塔;二异丁烯精制塔压力0.2mpa,塔顶温度:45℃;
固定床聚合反应温度90℃,反应压力0.5mpa;将固定床预聚反应的聚合反应产物冷却后部分循环回固定床聚合反应器进料口以稀释进料中异丁烯浓度并取走反应热,以重量份计循环量为混合c4进料的6倍;
催化蒸馏塔床层入口温度90℃,反应压力0.5mpa。
实施例2
采用如图1的催化蒸馏法工艺;
混合c4中含异丁烯18.5%,含正丁烯16.3%,进料量:300g/h;
固定床聚合反应器催化剂装量:300ml,催化剂为大孔磺酸阳离子交换树脂,交换容量为5.0mmol〔h+〕/g,其制备方法同实施例1中聚合催化剂的制备方法,区别是:向450g水中加入4g羟丙基甲基纤维素、0.3g聚丙烯酰胺、2g仲烷基磺酸钠,加热搅拌,使其完全溶解,形成均匀的水溶液。再加入由60g苯乙烯、40g二乙烯基甲苯、3克过氧化苯甲酰、40g液体石蜡、0.6g过氧化新癸酸叔丁酯组成的有机相混合物。
催化蒸馏塔催化剂装量:200ml;
原料中配入聚合反应抑制剂,抑制剂与新鲜进料中异丁烯的摩尔比为1.0:1;
聚合产品送二异丁烯精制塔;二异丁烯精制塔压力0.2mpa,塔顶温度:45℃;
固定床聚合反应温度30℃,反应压力1.6mpa;将固定床预聚反应的聚合反应产物冷却后部分循环回固定床聚合反应器进料口以稀释进料中异丁烯浓度并取走反应热,以重量份计循环量为混合c4进料的10倍;
催化蒸馏塔床层入口温度30℃,反应压力1.6mpa。
实施例3
采用如图1的催化蒸馏法工艺;
混合c4中含异丁烯18.5%,含正丁烯16.3%,进料量:300g/h;
固定床聚合反应器催化剂装量:400ml,催化剂为大孔磺酸阳离子交换树脂,交换容量为5.5mmol〔h+〕/g,其制备方法同实施例1中聚合催化剂的制备方法,区别是:向500g水中加入0.4g羟丙基甲基纤维素、6g聚丙烯酰胺、1g仲烷基磺酸钠,加热搅拌,使其完全溶解,形成均匀的水溶液。再加入由80g苯乙烯、30g二乙烯基甲苯、1.5克过氧化苯甲酰、33g正十二烷、4g过氧化新癸酸叔丁酯组成的有机相混合物;
催化蒸馏塔催化剂装量:200ml;
原料中配入聚合反应抑制剂,抑制剂与新鲜进料中异丁烯的摩尔比为1.5:1;
聚合产品送二异丁烯精制塔,二异丁烯精制塔压力0.2mpa,塔顶温度:45℃;
固定床聚合反应温度140℃,反应压力0.2mpa;将固定床预聚反应的聚合反应产物冷却后部分循环回固定床聚合反应器进料口以稀释进料中异丁烯浓度并取走反应热,以重量份计循环量为混合c4进料的9倍;
催化蒸馏塔床层入口温度55℃,反应压力0.6mpa。
实施例4
采用如图1的催化蒸馏法工艺;
混合c4中含异丁烯38.5%,含正丁烯1.63%,进料量:300g/h;
固定床聚合反应器催化剂装量:500ml,催化剂为大孔磺酸阳离子交换树脂,交换容量为4.0mmol〔h+〕/g;
催化蒸馏塔催化剂装量:200ml;
原料中配入聚合反应抑制剂,抑制剂与新鲜进料中异丁烯的摩尔比为0.003:11;
聚合产品送二异丁烯精制塔,二异丁烯精制塔压力0.2mpa,塔顶温度:45℃;
固定床聚合反应温度20℃,反应压力4.0mpa;将固定床预聚反应的聚合反应产物冷却后部分循环回固定床聚合反应器进料口以稀释进料中异丁烯浓度并取走反应热,以重量份计循环量为混合c4进料的10倍;
催化蒸馏塔床层入口温度25℃,反应压力36mpa。
对比例1~2的工艺同实施例1。
实施例1~4及对比例1~2为本发明在上述操作条件下添加不同抑制剂的结果。聚合产品经产品精制塔静置后二异丁烯的纯度均在98%以上。
实施例5~6及对比例3~4采用如图2串联固定床法间接烷基化工艺;
混合c4中含异丁烯48.5%,含正丁烯26.3%;
进料量:400g/h;
第一、二级固定床聚合反应器催化剂装量分别为:400ml、300ml,催化剂为大孔磺酸阳离子交换树脂,交换容量为5.0mmol〔h+〕/g;
两级反应器进料中分别配入适量聚合反应抑制剂,总抑制剂量与新鲜进料中的异丁烯的摩尔比为0.02∶1,第一级反应的加入量为总量的70%;
第一级固定床聚合反应温度50.0℃,反应压力1.0mpa;将第一级固定床预聚反应的聚合反应产物冷却后部分循环回第一级固定床聚合反应器进料口以稀释进料中异丁烯浓度并取走反应热,以重量份计循环量为混合c4进料的13倍;
第二级固定床聚合反应温度70.0℃,反应压力1.0mpa;
二异丁烯精制塔压力0.2mpa,塔顶温度:45℃。
实施例5~6及对比例3~4为本发明在上述操作条件下添加不同抑制剂的结果。聚合产品经产品精制塔静置后产品二异丁烯的纯度均在98%以上。
实施例7~8采用如图2串联固定床法间接烷基化工艺;
混合c4为mtbe分解产物经水洗除去甲醇、再经精馏精制后得到,其中含异丁烯99.99%,含正丁烯0.0003%;
进料量:450g/h
第一、二级固定床聚合反应器催化剂装量分别为:500ml、300ml,催化剂为大孔磺酸阳离子交换树脂,交换容量为5.2mmol〔h+〕/g;
两级反应器进料中分别配入适量聚合反应抑制剂,总抑制剂量与新鲜进料中的异丁烯的摩尔比为0.3∶1,第一级反应的加入量为总量的70%;
第一级固定床聚合反应温度40.0℃,反应压力1.0mpa;将第一级固定床预聚反应的聚合反应产物冷却后部分循环回第一级固定床聚合反应器进料口以稀释进料中异丁烯浓度并取走反应热,以重量份计循环量为混合c4进料的15倍;
第二级固定床聚合反应温度50.0℃,反应压力1.0mpa;
二异丁烯精制塔压力0.2mpa,塔顶温度:45℃。
实施例7~8为本发明在上述操作条件下添加不同抑制剂的结果。聚合产品经产品精制塔静置后产品二异丁烯的纯度均在98%以上。
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!