一种以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物及其制备方法与流程
本发明涉及噻吩衍生物及有机半导体材料领域,具体涉及一种以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物及其制备方法。
背景技术:
齐聚噻吩衍生物有机半导体因其具有优异的平面性、明确的分子结构、固定的分子量、易于纯化等优点,成为了有机半导体领域研究的重点。
稠环噻吩其结构具有较强的刚性平面共轭,若将其引入齐聚噻吩化合物的分子骨架中,可增强齐聚物分子间π-π堆积,提高载流子迁移效率。二噻吩并吡咯是一种常见的稠环类噻吩衍生物,分子结构中含有两个具有富电子体系的噻吩环,可在有机半导体材料中充当给电子单元,从而提升有机半导体性能。
此外,s,s-二氧-二苯并噻吩是一个平面型的受电子单元,并具有较高的电子亲和势。若将二噻吩并吡咯给电子结构单元与s,s-二氧-二苯并噻吩受电子单元组合,则可设计出一种新型给-受型齐聚噻吩衍生物材料,并有望在半导体器件领域获得应用。
技术实现要素:
本发明的目的在于提供一种以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物及其制备方法,以解决上述背景技术中提出的问题。
为实现上述目的本发明采用以下技术方案:
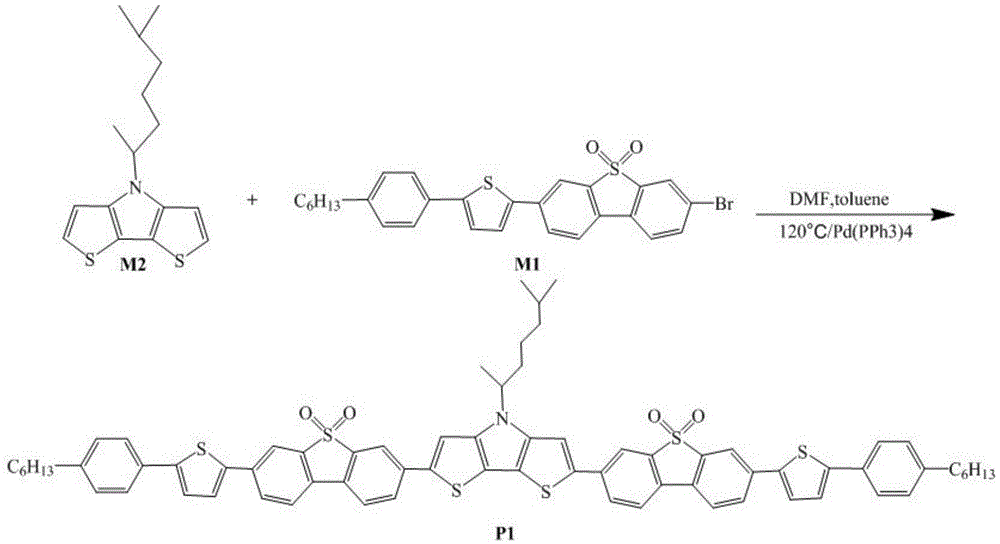
一种以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物,其化学结构式如下p1所示:
一种以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物的制备方法,包括如下步骤:
在惰性气氛中,将中间体3-溴-7-(5-己基-2,2’-二噻吩基)二苯并噻吩-s,s-二氧化物与中间体2,6-二(三甲基锡)-n-异辛烷基二噻吩并[3,2-b:2',3'-d]吡咯以及催化剂溶于溶剂中进行stille交叉偶联反应,制备得到所述以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物(合成流程图如图1所示)。
作为本发明进一步的方案,所述stille交叉偶联反应温度为100-120℃,反应时间为12-72小时。
作为本发明进一步的方案,中间体3-溴-7-(5-己基-2,2’-二噻吩基)二苯并噻吩-s,s-二氧化物(m1)与中间体2,6-二(三甲基锡)-n-异辛烷基二噻吩并吡咯(m2)质量摩尔比为1.5-3:1。
作为本发明进一步的方案,stille交叉偶联反应在氮气或氩气氛围下进行。
作为本发明进一步的方案,所述stille交叉偶联反应所用催化剂为四(三苯基膦)钯。
作为本发明进一步的方案,所述stille交叉偶联反应所用溶剂为无水n,n-二甲基甲酰胺(dmf)或无水甲苯。
作为本发明进一步的方案,还包括以下步骤,反应所得终产物的粗产物先进行萃取,再进行梯度柱层析洗脱,最后用沉淀法提纯。
萃取所用溶剂为二氯甲烷,柱层析所用洗脱剂为石油醚和二氯甲烷的混合液,沉淀法提纯所用溶剂为四氯乙烷,沉淀剂为甲醇。
本发明的有益效果是:本发明涉及的一种以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物,经过光物理性能和电化学性能测定,发现具有以下性能:1)合适的光学带隙,egopt(ev)=-2.24ev。2)较低的homo能级。其homo能级为-5.48ev,lomo能级为-3.04ev,电化学能隙(egec)为-2.44ev,与uv-vis计算的光学带隙(-2.24ev)比较接近。3)相比在二氯甲烷溶液中,该噻吩衍生齐聚物在固体薄膜中的紫外吸收光谱和光致发光光谱均发生了一定波长的红移,分别为5nm和116nm。证明该齐聚物分子间存在一定的π-π相互作用。4)该齐聚物的起始熔点为251.93℃,熔点为268.37℃。当温度升至300℃时,未发现氧化和分解,表明齐聚物具有优异的热稳定性。
因此,以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物dtp(dbtotph)2是一种有望应用于有机发光二极管(oled)和有机太阳能电池(osc)器件制备的新材料。
附图说明
图1为本发明以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物的合成流程图。
图2为以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物的紫外吸收(uv-vis)光谱图。
图3为以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物的光致发光(pl)光谱图。
图4为以二噻吩并吡咯为给电子中心的齐聚噻吩衍生物的差示扫描量热(dsc)曲线图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整的阐述。
实施例1
在250ml的烧瓶中,依次加入2,6-二(三甲基锡)-n-异辛烷基二噻吩并[3,2-b:2',3'-d]吡咯(0.62g,1.00mmol)、3-溴-7-(2-正己基苯基-噻吩)-s,s-二氧-二苯并噻吩(1.08g,2.0mmol)、和四(三苯基膦)钯(0.17g,0.15mmol),溶于100ml干燥的甲苯中;氮气保护下,在110℃下连续搅拌反应60h;将反应混合物冷却至室温,加入蒸馏水,用二氯甲烷萃取三次;合并有机相,依次经无水硫酸镁干燥、过滤、减压蒸馏去除溶剂,剩余物采用硅胶柱层析阶梯洗脱,洗脱剂为二氯甲烷与石油醚混合液。然后将产物溶于四氯乙烷,以甲醇为沉淀剂进行提纯。最后得到褐色固体0.31g,产率为25%。
实施例2
在250ml的烧瓶中,依次加入2,6-二(三甲基锡)-n-异辛烷基二噻吩并[3,2-b:2',3'-d]吡咯(0.50g,0.80mmol)、3-溴-7-(2-正己基苯基-噻吩)-s,s-二氧-二苯并噻吩(0.86g,1.6mmol)、和四(三苯基膦)钯(0.14g,0.12mmol),溶于80ml干燥的n,n-二甲基甲酰胺中;氮气保护下,在100℃下连续搅拌反应72h;将反应混合物冷却至室温,加入蒸馏水,用二氯甲烷萃取三次;合并有机相,依次经无水硫酸镁干燥、过滤、减压蒸馏去除溶剂,剩余物采用硅胶柱层析纯化,洗脱剂为二氯甲烷与石油醚混合液。然后将产物溶于四氯乙烷,以甲醇为沉淀剂进行提纯。最后得到褐色固体0.28g,产率为29%。
实施例3
在250ml的烧瓶中,依次加入2,6-二(三甲基锡)-n-异辛烷基二噻吩并[3,2-b:2',3'-d]吡咯(0.37g,0.60mmol)、3-溴-7-(2-正己基苯基-噻吩)-s,s-二氧-二苯并噻吩(0.65g,1.2mmol)、和四(三苯基膦)钯(0.1g,0.09mmol),溶于70ml干燥的n,n-二甲基甲酰胺中;氮气保护下,在120℃下连续搅拌反应48h;将反应混合物冷却至室温,加入蒸馏水,用二氯甲烷萃取三次;合并有机相,依次经无水硫酸镁干燥、过滤、减压蒸馏去除溶剂,剩余物采用硅胶柱层析纯化,洗脱剂为二氯甲烷与石油醚混合液。然后将产物溶于四氯乙烷,以甲醇为沉淀剂进行提纯。最后得到褐色固体0.20g,产率为27.7%。
采用核磁共振(nmr)对所合成齐聚物结构进行表征所得数据如下:
1hnmr(cdcl3,400hz,δ/ppm):8.38(d,3jh-h=10.8hz,2h,dbto-h),8.25(t,3jh-h=8.6hz,2h,dbto-h),8.07(d,3jh-h=10.8hz,4h,dbto-h),7.87(d,3jh-h=16hz,4h,dbto-h),7.73(m,4h,p-h),7.54(m,4h,p-h),7.47(d,3jh-h=8hz,4h,t-h),7.26(s,2h,dtp-h),4.31(t,3jh-h=6.8hz,h,n-ch),2.68(d,3jh-h=4.8hz,4h,p-ch2),1.74~1.26(m,26h,ch2ch3),0.99~0.83(m,12h,ch3).13cnmr(c2cl4d2,100hz,δ/ppm)δ147.04,142.04,140.73,138.69,137.87,134.51,132.63,128.47,122.74,122.44,122.02,120.48,54.00,36.91,35.73,33.31,31.82,29.92,27.13,22.86,22.49,21.84,14.19。
以上所述为本发明较佳实施例,对于本领域的普通技术人员而言,根据本发明的教导,在不脱离本发明的原理与精神的情况下,对实施方式所进行的改变、修改、替换和变型仍落入本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!