光致收缩金属有机框架化合物的制作方法
本发明涉及光敏材料技术领域,尤其涉及一种光致收缩金属有机框架化合物,其同分异构体及制备方法。
背景技术:
固态柔性材料因其是一种对外部刺激(例如光,电/磁场,热,ph和湿度)在分子水平有机械响应的材料,故而在智能器件、记忆存储和药物释放等方面有着巨大的应用前景。为了提升柔性材料的性能,了解这类材料的结构-功能之间的关系是固态化学的一个重要研究领域,因而越来越多的科学家投身于此。通常来说,外部刺激势必引起这类柔性材料微观结构的变化,而这种结构的变化主要在于主体结构框架内化学键的振动、断裂/重组、有机刚性基团的旋转(例如芳香环)以及“铰链运动(hinge-like)”等等。热膨胀作为一个具有代表性的例子,近来已在沸石、金属氰化物、金属氧化物、金属有机框架等柔性材料被广泛研究报道。目前,在自然界发现的材料中存在三种截然不同的膨胀材料。一,正膨胀,也就是人们所熟知的热胀冷缩现象,即材料的体积随热流的增加而增加;二,零膨胀,即材料的体积不随热流的变化而变化;三,负膨胀,可简单理解为热缩冷胀现象,即材料体积随热流增加而减小。在这其中零膨胀与负膨胀材料在航空航天材料,精密器件方面显得尤为重要。但是,随着社会的发展,热致膨胀材料,尤其是负膨胀,零膨胀材料已不能满足科学技术的需求。
作为固态柔性材料的一个分支,光响应固态材料因其独特的特性得到了研究者的广泛关注。一方面,光作为清洁能源,对环境造成污染。另一方面,光响应材料以一种无损伤、非接触的方式来引发。由于光响应材料的性能与其结晶度、自由体积和光敏分子的取向有这很大的关联,使得在“自下而上”设计合成具有高灵敏度、快速响应的各向异性光致动材料方面仍然存在很大的挑战。基于此,masahiroirie课题组报道了光照下二芳基乙烯生色团衍生物分子晶形状快速可逆变化的例子(参见:kobatake,s.;takami,s.;muto,h.;ishikawa,t.;irie,m.nature2007,446,778-781),正方形二乙烯基取代物(1,2-bis(2-ethyl-5-phenyl-3-thienyl)perfluorocyclopentene)的单晶在紫外光照下发生周环反应,变成菱形状单晶,并伴随着颜色由无色到蓝色的变化。棒状的单晶可朝紫外光照射的方向弯曲,并在可见光照射下恢复原形状。将其应用于显微镜物体的移动中,显示出快速高效的光能转化。由此可见,这类光控收缩或记忆材料在光响应器件和分子机械驱动器方面有着重要的应用前景。
技术实现要素:
本发明要解决的技术问题是提供一种新的光致收缩金属有机框架化合物,该有机框架化合物制备方法简单,反应条件温和,且光转化速率快。
为了解决上述技术问题,本发明提供了一种光致收缩金属有机框架化合物,所述光致收缩金属有机框架化合物的分子式为c44h36n2o9zn2,化学式为[zn2(bdc)2(3-ch3-spy)]·h2o(在下文中以zn2-1表示),该金属有机框架化合物为无色棒状晶体,其晶体学参数为:
(1)晶系:单斜晶系;
(2)空间群:p21/c;
(3)
(4)dc=1.425/g·cm–3,z=4,μ=1.268(mo-kα)/mm–1;
(5)总衍射点数:60722,独立衍射点数:9099;
(6)f(000)=1744,r1a=0.0691,wr2b=0.1908,gofc=0.972。
其中,bdc代表1,4-对苯二甲酸根,3-ch3-spy代表3-甲基苯乙烯基-4-吡啶,1,4-对苯二甲酸根与3-甲基苯乙烯基-4-吡啶的化学结构分别如式(ⅱ)、(ⅲ)所示:
此外,本发明还提供了上述光致收缩金属有机框架化合物的制备方法,包括如下步骤:
将七水合硫酸锌或硝酸锌、1,4-对苯二甲酸或其水溶盐和3-甲基苯乙烯基-4-吡啶溶于n,n’-二甲基甲酰胺和去离子水的混合溶剂中,在135~155℃下反应5~12h,得到所述光致收缩金属有机框架化合物。
作为优选的方案,所述水合硫酸锌或硝酸锌、1,4-对苯二甲酸或其水溶盐和3-甲基苯乙烯基-4-吡啶的摩尔比为1~2:1:1,更优选的为1:1:1。
作为优选的方案,所述反应的原料为七水合硫酸锌、1,4-对苯二甲酸和3-甲基苯乙烯基-4-吡啶。
作为优选的方案,所述反应温度为140~150℃,反应时间为6~8h。更优选的,所述反应温度为145℃,反应时间为8h。
作为优选的方案,所述混合溶剂中n,n’-二甲基甲酰胺和去离子水的体积比为1:1~4,更优选的为2:3。
本发明得到的金属有机框架化合物[zn2(bdc)2(3-ch3-spy)]·h2o(zn2-1)具有光致收缩性,该化合物经过420nm~365nm波长的光源照射,能够得到一系列同分异构体化合物。该同分异构体化合物的分子式为c44h34n2o8zn2,化学通式为[zn2(bdc)2(rctt-tcdp)](下文中以zn2-2n表示)。
其中,rctt-tcdp代表不同构型1,3-二(4-吡啶)-2,4-二间甲苯基环丁烷,1,3-二(4-吡啶)-2,4-二吡啶基环丁烷化学结构如式(ⅰ)所示:
上述制备方法中,当光源照射时,金属有机框架化合物zn2-1内一对3-甲基苯乙烯基-4-吡啶发生[2+2]环加成反应,生成式(ⅰ)所示的环丁烷衍生物,双齿环丁烷衍生物通过连接两个层状的[zn2(bdc)2]n网状结构,形成一个二重穿插的三维金属有机框架化合物(具体过程参见附图8)。
作为优选的方案,所述光源选自波长为420nm、405nm、385nm或365nm的光源,得到的化学式为[zn2(bdc)2(rctt-tcdp)]的金属有机框架化合物,并将这种材料分别命名为zn2-2a,zn2-2b,zn2-2c和zn2-2d。
此外,本发明还提供了一种运用原子力显微镜(afm)表征晶体材料宏观收缩的方法。其包括下列步骤:挑选单颗表面无明显缺陷、且光滑规则的单晶zn2-1,并将其置于洁净的载玻片中间。用原子力显微镜(afm)测试并记录该晶体在385nm波长led光源光照前后晶体的表面形貌。分析光照前后晶体表面形貌发现,光照后由于[2+2]环加成微观重组,导致宏观晶体表面产生“褶皱”形貌,从而将化合物的微观与宏观联系起来。利用原子力显微镜(afm)的方法来研究晶体宏观与微观之间的联系,测试方便简单、实验结果直接、明显。
此外,本发明还提供了一种如式(ⅰ)所示的环丁烷衍生物的制备方法,该制备方法包括:
提供光致收缩金属有机框架化合物zn2-1,经420nm~365nm波长的光源照射以发生环加成反应,得到的产物经强酸处理,中和,萃取后,即得式(ⅰ)所示的环丁烷衍生物。
作为优选的方案,所述强酸选自浓盐酸、硝酸、硫酸中的至少一种,更优选的为浓盐酸。
作为优选的方案,得到的产物使用1mol/l的氢氧化钠中和至ph=7,再采用二氯甲烷萃取,即得具有顺反构象的环丁烷衍生物。
本发明的有益效果在于:
1、本发明制备得到了一种新的金属有机框架化合物,该化合物在不同波长光的照射下能发生[2+2]环加成反应,并得到一系列各向异性收缩的同分异构体化合物。所述金属有机框架化合物的制备方法简单,反应条件温和,且光转化速率快,能够准确调节材料的收缩性能。
2、本发明中光致收缩的手段为非接触型和损伤型,体积调控精准度高,且整个调控过程无任何化学试剂参与,安全可靠。
3、本发明利用强酸对含环丁烷衍生物的金属有机框架化合物(zn2-2n)的刻蚀作用,获得相应的环丁烷衍生物,通过简单的搅拌、中和、萃取分离、干燥,可以得到纯净的产物,反应转化率高,且刻蚀后h2bdc还可以回收利用,符合绿色化学理念。
附图说明
附图1为实施例一中化合物[zn2(bdc)2(3-ch3-spy)]·h2o的合成示意图;
附图2为实施例一中化合物[zn2(bdc)2(3-ch3-spy)]·h2o的三维堆积图;
附图3为实施例一中化合物[zn2(bdc)2(3-ch3-spy)]·h2o的x-射线粉末衍射图;
附图4为实施例一中化合物[zn2(bdc)2(3-ch3-spy)]·h2o的热重分析图;
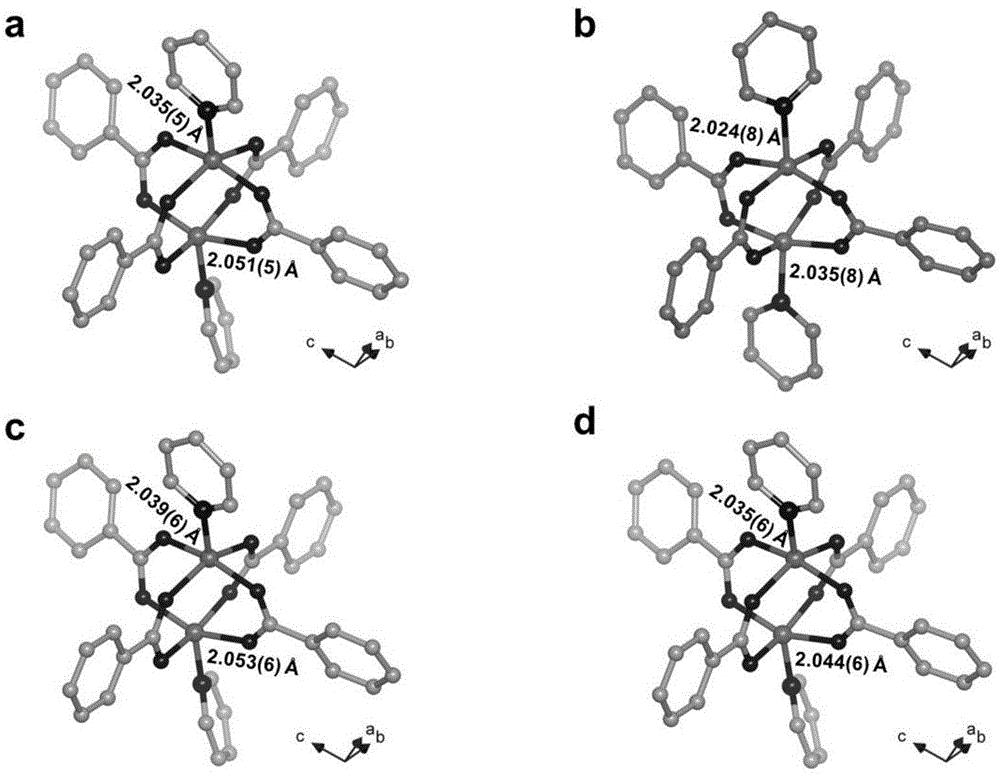
附图5为实施例二中化合物[zn2(bdc)2(rctt-tcdp)](zn2-2a)的三维结构图;
附图6为实施例五中化合物[zn2(bdc)2(rctt-tcdp)](zn2-2d)的x-射线粉末衍射图;
附图7为实施例五中化合物[zn2(bdc)2(rctt-tcdp)](zn2-2d)的热重分析图;
附图8为实施例六中光致收缩示意图;
附图9为实施例六中光致收缩装置图;
附图10为实施例六中光致晶格参数b轴和c轴分布图;
附图11为实施例六中光致晶格参数a轴和体积v分布图;
附图12为实施例六中光致“轮浆式(paddle-wheel)”部分结构变化图;
附图13为实施例六中光致环丁烷衍生物结构变化图;
附图14为实施例七中光照前后晶体表面的原子力显微镜(afm)图;
附图15为实施例七中光照前后晶体的尺寸大小光学显微图;
附图16为实施例八中3-ch3-spy和环丁烷衍生物rctt-tcdp的1hnmr图谱;
附图17为实施例八中环丁烷衍生物rctt-tcdp的13cnmr图谱;
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
实施例一:金属有机框架化合物[zn2(bdc)2(3-ch3-spy)]·h2o的制备
将七水合硫酸锌(143mg,0.5mmol)、3-甲基苯乙烯基-4-吡啶(97mg,0.5mmol)和1,4-对苯二酸(83mg,0.5mmol)的混合物装入到25ml的厚壁耐压瓶中,加入10ml体积比为2:3的n,n’-二甲基甲酰胺和去离子水的混合溶剂。拧紧瓶盖,超声分散10分钟。然后置于程序升温至145℃的烘箱中加热8小时。自然冷却至室温,得到无色棒状的晶体[zn2(bdc)2(3-ch3-spy)]·h2o(zn2-1)。用乙醇洗涤收集,然后置于60℃烘箱干燥,产率:138.2mg(63%,基于3-甲基苯乙烯基-4-吡啶计算)。
元素分析(%):c44h36n2o9zn2;理论值:c60.92,h4.18,n3.23;实测值:c61.3,h4.21,n3.31。
红外光谱(溴化钾压片法):3443(w),3031(w),2920(w),1946(w),1641(s),1502(m),1386(s),1224(w),1032(m),955(m),869(m),823(s),746(s),688(m),561(m)cm-1。
对该化合物进行了x-射线单晶衍射表征,x-射线粉末衍射和热分析。其晶体学参数见表1,x-射线粉末衍射图和热分析图参见附图3,4。
表1实施例一的金属有机框架化合物的晶体学参数
附图1-2示出了zn2-1化合物的合成示意图和空间结构示意图。从图中可以看出,上述含烯烃配体的金属有机框架的中心金属离子为zn2+,其通过“轮浆式”的[zn2(coo)4]为次级构筑单元,与四个bdc相互连接成一个二维的网状结构。这些二维结构在轴向方向与两个3-ch3-spy配体相连接形成一个二维的层状结构,紧接着通过烯烃配体之间的π-π作用进一步沿b轴堆积为一个三维的超分子框架。此外,该金属有机框架化合物为棒状晶态,结晶为单斜p21/c晶系。
实施例二:金属有机框架化合物[zn2(bdc)2(rctt-tcdp)](zn2-2a)的制备
室温下,取少量的zn2-1晶体于干净的载玻片中,保持光源和晶体的距离为2cm的情况下,用420nm波长的led灯照射30分钟,获得100%转化的[2+2]环加成产物[zn2(bdc)2(rctt-tcdp)](zn2-2a)。
元素分析(%):c44h34n2o8zn2,理论值:c62.21,h4.03,n3.30;实测值:c61.73,h4.16,n3.28。
红外光谱(溴化钾压片法):3440(w),3066(w),2938(w),2359(w),1938(w),1824(w),1640(s),1504(m),1393(s),1223(w),1032(m),919(w),881(m),821(s),745(s),688(m),546(s)cm-1。
对该产物进行x-射线单晶衍射测试。其晶体学参数见表2,单晶结构图参见附图5。
实施例三:金属有机框架化合物[zn2(bdc)2(rctt-tcdp)](zn2-2b)的制备
室温下,取少量的zn2-1晶体于干净的载玻片中,保持光源和晶体的距离为2cm的情况下,用405nm波长的led灯照射30分钟,获得100%转化的[2+2]环加成产物[zn2(bdc)2(rctt-tcdp)](zn2-2b)。
元素分析(%):c44h34n2o8zn2,理论值:c62.21,h4.03,n3.30;实测值:63.09,h4.11,n3.39。
红外光谱(溴化钾压片法):3439(w),3023(w),2936(w),1940(w),1823(w),1640(s),1504(m),1393(s),1223(w),1032(m),952(w),882(m),822(s),744(s),706(m),545(s)cm-1。
对该产物进行x-射线单晶衍射测试,其晶体学参数见表2。
实施例四:金属有机框架化合物[zn2(bdc)2(rctt-tcdp)](zn2-2c)的制备
室温下,取少量的zn2-1晶体于干净的载玻片中,保持光源和晶体的距离为2cm的情况下,用385nm波长的led灯照射30分钟,获得100%转化的[2+2]环加成产物[zn2(bdc)2(rctt-tcdp)](zn2-2c)。
元素分析(%):c44h34n2o8zn2,理论值:c62.21,h4.03,n3.30;实测值:c63.34,h4.35,n3.21。
红外光谱(溴化钾压片法):3441(w),3034(w),2935(w),2344(w),1938(w),1830(w),1640(s),1502(m),1388(s),1224(w),1018(m),919(w),881(m),821(s),745(s),688(m),545(s)cm-1。
对该产物进行x-射线单晶衍射测试,其晶体学参数见表2。
实施例五:金属有机框架化合物[zn2(bdc)2(rctt-tcdp)](zn2-2d)的制备
室温下,取少量的zn2-1晶体于干净的载玻片中,保持光源和晶体的距离为2cm的情况下,用365nm波长的led灯照射30分钟,获得100%转化的[2+2]环加成产物[zn2(bdc)2(rctt-tcdp)](zn2-2d)。
元素分析(%):c44h34n2o8zn2,理论值:c62.21,h4.03,n3.30;实测值:c63.17,h4.14,n3.27。
红外光谱(溴化钾压片法):3439(w),3058(w),2938(w),2359(w),1938(w),1824(w),1640(s),1504(m),1393(s),1223(w),1032(m),919(w),841(s),807(s),744(s),716(ms),546(s)cm-1。
对该产物进行x-射线单晶衍射测试,x-射线粉末衍射和热分析。其晶体学参数见表2,x-射线粉末衍射图和热分析图参见附图6,7。
表2实施例二-五的金属有机框架化合物的晶体学参数
实施例六:光致非线性收缩行为
与实施例二至例五的方法相同,采用不同波长光(420nm、405nm、385nm、365nm)来照射实施例一中的金属有机框架化合物,得到各向异性收缩的一系列金属有机框架化合物[zn2(bdc)2(rctt-tcdp)](zn2-2n,n=a-d),且这些化合物的晶格参数呈现非线性分布。光致收缩示意图和装置图参见附图8,9。
如附图10所示,在用420nm~365nm波长的光照射后,晶体的晶格参数呈现出不同的变化趋势。其中,b轴、c轴晶格参数基本保持不变;a轴和晶格体积v呈现出非线性收缩。且在385nm光照射下,a轴和晶格体积v达到最大幅度的收缩,分别为7.33%和6.18%。通过x-射线单晶衍射分析其单晶结构发现,这种光致非线性收缩现象主要因为化合物对光子吸收转化的差异,导致在光照后化合物结构发生不同程度的变化(参见附图12,13)。基于实验测试数据,用拟合的公式(1)、(2)表示a轴和晶格体积v与光照波长(λ)之间的关系:
a=(ω1’+ω2’λ+ω3’λ2)-1公式(1)
ω1’、ω2’和ω3’的数值分别为
v=(ω1+ω2λ+ω3λ2)-1公式(2)
ω1’、ω2’和ω3’的数值分别为
实施例七:光致晶体表面的变化与表征
挑选单颗表面无明显缺陷、且光滑规则的单晶zn2-1,尺寸大小约为2×2×4mm,并将其置于洁净的载玻片中间。首先,用原子力显微镜(afm)测试并记录该晶体表面的形貌。然后,将该晶体在与保持光源的距离为2cm的情况下,用385nm波长的led灯照射10分钟,并同样地用原子力显微镜(afm)测试并记录光照后该晶体表面的形貌。
如附图14所示,光照前,晶体表面光滑,界面厚度为4.1nm。在光照后,由于光反应的发生,晶体表面重组,“褶皱”状的形貌存在于晶体表面,界面厚度达到了143.4nm。因此,afm图提供了光照下晶体宏观收缩于微观结构转变的直接证据。与此同时,棒状的晶体在光照下发生了明显的收缩(参见附图15)。
实施例八:环丁烷衍生物rctt-tcdp的合成与表征
取200mg化合物zn2-2d于烧瓶中,加10ml浓盐酸搅拌24小时。用1mol/l的氢氧化钠中和至ph=7,继续加入20ml二氯甲烷搅拌3小时。旋转蒸发后获得浅黄色粉末rctt-tcdp,产率为92%。rctt-tcdp的1hnmr与13cnmr图谱参见附图16,17。
1hnmr(400mhz,cdcl3):δ(ppm)8.35(s,4h,py),7.07–7.04(t,2h,phenyl),7.04–7.01(d,4h,py),7.00–6.88(m,4h,phenyl),4.43(dd,j=18.9,6.1hz,4h,cyclobutane),2.22(s,6h,methyl);
13cnmr(75mhz,cdcl3):δ(ppm)149.75,149.06,138.77,137.96,128.57,128.29,127.52,124.76,123.34,77.29,77.08,76.87,46.75,46.55,21.37。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!