一种高刻蚀对比度的嵌段共聚物及其制备和应用的制作方法
本发明涉及高分子材料领域,尤其涉及一种高刻蚀对比度的嵌段共聚物及其制备和应用。
背景技术:
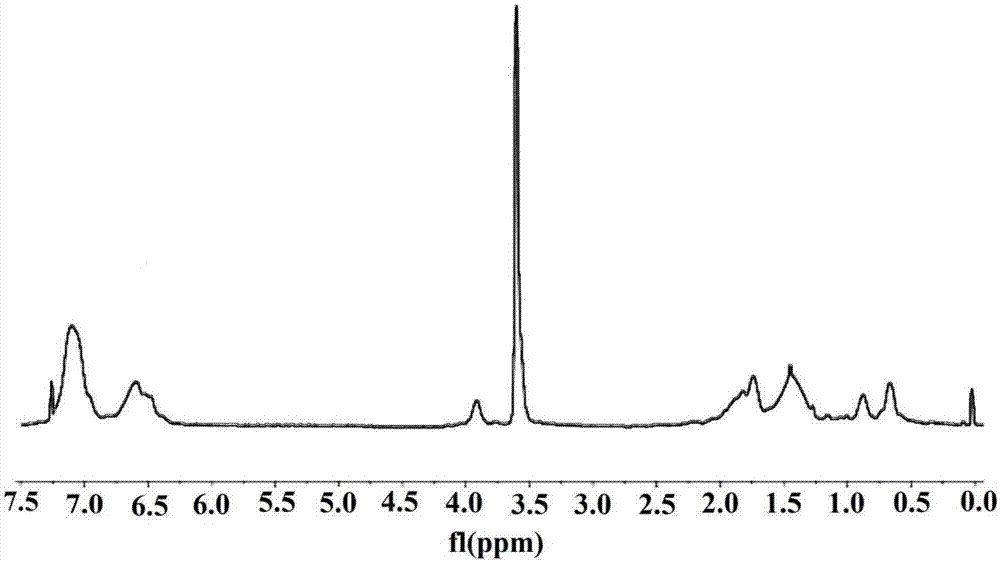
:过去的50年内,半导体产业一直沿着摩尔定律前行,光刻材料的分辨率也不断提高,然而,现有的193nm浸没式光刻技术已基本到达了其分辨率的极限。对于sub-10nm的光刻,工业上普遍认可的有两种技术,即极紫外光刻(euvl)技术以及导向自组装(dsa)技术,其中dsa技术以其过程简单、成本低廉而备受关注。dsa技术利用嵌段高分子的微相分离特性,从而实现高分辨率的有序纳米级图案。传统的作法是将具有自组装特性的嵌段高分子旋涂在一定的模板内(通常通过193nm曝光制成),通过侧壁或者基底的导向作用,得到更高分辨率的周期性图案结构。传统的dsa材料聚苯乙烯-聚甲基丙烯酸酯嵌段聚合物(ps-b-pmma)是工业界和学术界研究最广泛的dsa材料。然而,ps-b-pmma材料分辨率极限为22nm(full-pitch),限制了其在sub-10nm图案化的应用,并且ps-b-pmma材料的刻蚀对比度较差,限制了其图案转移,而不利于后期应用。综上所述,本领域急需开发一种具有高刻蚀对比度的嵌段共聚物。技术实现要素:本发明技术方案所要解决的技术问题是提供一种高刻蚀对比度的嵌段共聚物,该嵌段共聚物可在加热退火后自组装,实现高分辨率的相分离和图案化,分子结构中引入的金刚烷或其衍生物属于高碳原子密度的基团,能够大大增加其刻蚀对比度。为解决上述技术问题,本发明一方面提供了一种高刻蚀对比度的嵌段共聚物,包含嵌段a和嵌段b,所述嵌段a由以下结构式i的单体聚合得到:其中,r1、r2选自氢、由c1-c10烷基取代或者未取代的丙烯酸酯基、取代或未取代的c3-c6环烷酯基、取代或未取代的c6-c10芳香酯基、取代或未取代的含1-3个选自n、o、s的c6-c10的杂芳酯基、取代或未取代的苯乙烯基,且r1与r2不同时为氢并且不同时包含双键;r3选自氢、c1-c10的烷基、取代或未取代的c3-c6环烷基、取代或未取代的c6-c10芳香基、取代或未取代的含1-3个选自n、o、s的c6-c10的杂芳基、卤素;r4选自氢、氰基、c1-c10的烷基、卤素;r5选自氢、氰基、n、o、s、n为0-10的自然数;l1选自单键、取代或未取代的c1-c10的烷基、取代或未取代的c1-c6环酯基,所述取代指被选自下组的一个或多个取代基取代:羰基、酯基、羟基、卤素。本发明一优选实施例,r1选自氢、丙烯酸酯基或者由c1-c10烷基取代的丙烯酸酯基;r2选自氢、取代或未取代的c6-c10的苯乙烯基;r3选自氢、c1-c10的烷基、乙酸乙酯基、羧基、羟基。本发明一优选实施例,所述嵌段a由选自下组的单体聚合得到:本发明一优选实施例,所述嵌段b由选自下组的单体聚合得到:取代或未取代的c3-c6烯基或其组合;其中,r1选自下组:无、取代或未取代的含1-5个si的硅烷基、取代或未取代的含1-5个ge的锗烷基、取代或未取代的含1-5个sn的锡烷基、取代或未取代的c1-c10的烷基、取代或未取代的c1-c6的烷氧基、取代或未取代的c3-c6的环烷基、取代或未取代的c3-c6的环烷基氧基、取代或未取代的c6-c10的芳基、取代或未取代的含1-3个选自n、o、s的c6-c10的杂芳基、羟基、卤素;其中,所述取代指被选自下组的一个或多个取代基取代:c1-c6烷基、含1-5个si的硅烷基、c1-c6烷氧基取代的含1-5个si的硅烷基、含1-5个si的硅烷基氧基、含1-5个si的硅烷基氧基取代的含1-5个si的硅烷基氧基、c1-c6烷氧基、羟基;r1的个数为0、1、2、3、4或5个;r2选自下组:无、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷氧基、羟基、卤素;所述取代指被选自下组的一个或多个取代基取代:卤素、羟基;r3选自下组:取代的c1-c10烷基、取代或未取代的c3-c6环烷基、取代或未取代的c6-c10芳基、取代或未取代的含1-3个选自n、o、s的c6-c10的杂芳基、取代或未取代的含1-5个si的硅烷基、取代或未取代的含1-5个ge的锗烷基、取代或未取代的含1-5个sn的锡烷基;所述取代指被选自下组的一个或多个取代基取代:c1-c6烷基、含1-5个si的硅烷基、c1-c6烷基取代的含1-5个si的硅烷基、c1-c6烷基取代的含1-5个si的硅烷基氧基、含1-5个si的硅烷基氧基取代的含1-5个si的硅烷基、c1-c6烷氧基取代的含1-5个si的硅烷基、含1-5个si的硅烷基氧基、c1-c6烷基取代的含4-10个si的笼状硅氧烷基;在所述取代或未取代的c3-c6烯基中,所述取代指被选自下组的一个或多个取代基取代:含1-5个si的硅烷基氧基、含1-5个si的硅烷基、c1-c6烷基取代的含1-5个si的硅烷基、c1-c6烷氧基取代的含1-5个si的硅烷基、c6-c10芳基取代的含1-5个si的硅烷基。本发明一优选实施方式,所述嵌段共聚物具有(a)m-(b)n两嵌段结构或(b)n1-(a)m-(b)n2三嵌段结构。本发明一优选实施方式,所述嵌段共聚物具有选自下组的特征:m/n=0.2-5或者m/(n1+n2)=0.2-5。本发明一优选实施方式,所述嵌段共聚物具有选自下组的一个或多个特征:1)所述嵌段共聚物的多分散性pdi≤2;2)所述嵌段共聚物的数均分子量为1000-120000;3)所述嵌段共聚物自组装所得产物的组装间距≤30nm。本发明一优选实施方式,所述嵌段共聚物选自下组:本发明的嵌段聚合物可通过阴离子聚合、氮氧自由基聚合,或其他活性自由基聚合制备而成。且本发明的嵌段聚合物不局限于线性的嵌段共聚物,可以为星型嵌段聚合物,接枝共聚物等。本发明的第二方面,提供了一种嵌段高分子材料,所述嵌段高分子材料包含本发明第一方面所述嵌段共聚物或由本发明第一方面所述嵌段共聚物制成。本发明的第三方面,提供了本发明第一方面所述嵌段共聚物的用途,用于制备选自下组的物质:dsa导向自组装材料、纳米催化剂、功能化纳米电子器件、便携式精密储存材料、生物医用纳米器件。与现有技术相比,本发明技术方案的高刻蚀对比度的嵌段共聚物具有以下优点:引入了嵌段a,大大增加了嵌段聚合物的刻蚀对比度,使得嵌段共聚物在加热退火后自组装,实现了高分辨率的相分离和图案化。附图说明图1为ps-b-pcm1的核磁共振氢谱图;图2为pmma-b-pcm2的核磁共振氢谱图;图3为pcm3-b-ppdfma的核磁共振氢谱图;图4为ptmss-b-pcm4的核磁共振氢谱图;图5为pcm5-b-phfmba的核磁共振氢谱图;图6为ps-b-pcm1退火后的saxs图;图7为pmma-b-pcm2退火后的saxs图;图8为pcm3-b-ppdfma退火后的saxs图;图9为ptmss-b-pcm4退火后的saxs图;图10为pcm5-b-phfmba退火后的saxs图.下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。具体实施方式实施例1嵌段共聚物1(ps-b-pcm1)的聚合以阴离子聚合为例,其制备步骤如下:将2~2.5ml的苯乙烯和30~35ml的四氢呋喃用二丁基镁溶液(1m,溶剂为正己烷)在40℃下处理0.5h,转移至反应瓶中。将反应瓶恢复至室温,搅拌均匀,然后置于-80℃的冷浴中,冷却15min。加入0.35~0.5mlsec-buli(1.3m,溶剂为正己烷),保持-80℃反应15min。将干燥好的金刚烷取代的丙烯酸酯类单体cm1(1.5~2ml)的温度降至-60℃,滴入反应体系,保持-80℃反应40min。产物在乙醇中沉淀,得到白色固体3.5~4.5g。共聚单体苯乙烯(ps)、cm1及嵌段共聚物1的结构式分别如下:及实施例2嵌段共聚物4(pmma-b-pcm2)的聚合以阴离子聚合为例,其制备步骤如下:将1.5~2ml甲基丙烯酸酯和30~35ml正己烷用二丁基镁溶液(1m,溶剂为正己烷)在40℃下处理0.5h,转移至反应瓶中。将反应瓶恢复至室温,搅拌均匀,然后置于-85℃的冷浴中,冷却15min。加入0.35~0.5mlsec-buli(1.3m,溶剂为正己烷),保持-85℃反应15min。将干燥好的金刚烷取代的丙烯酸酯类单体cm2(1.5~2ml)的温度降至-70℃,滴入反应体系,保持-85℃反应40min。产物在乙醇中沉淀,得到白色固体3.5~4.5g。共聚单体甲基丙烯酸酯(pmma)、cm2及嵌段共聚物4的结构式分别如下:及实施例3嵌段共聚物3(pcm3-b-ppdfma)的聚合以阴离子聚合为例,其制备步骤如下:将2~2.5ml金刚烷取代的丙烯酸酯类单体cm3和30~35ml正己烷用二丁基镁溶液(1m,溶剂为正己烷)在40℃下处理0.5h,转移至反应瓶中。将反应瓶恢复至室温,搅拌均匀,然后置于-80℃的冷浴中,冷却15min。加入0.35~0.5mlsec-buli(1.3m,溶剂为正己烷),保持-80℃反应15min。将干燥好的含氟丙烯酸酯类单体(1.5~2ml)的温度降至-60℃,滴入反应体系,保持-80℃反应40min。产物在乙醇中沉淀,得到白色固体2.5~4g。共聚单体含氟丙烯酸酯(ppdfma)单体、cm3及嵌段共聚物3的结构式分别如下:cm3和实施例4嵌段共聚物2(ptmss-b-pcm4)的聚合以阴离子聚合为例,其制备步骤如下:将2~2.5ml三甲基硅取代苯乙烯和30~35ml四氢呋喃用二丁基镁溶液(1m,溶剂为正己烷)在40℃下处理0.5h,转移至反应瓶中。将反应瓶恢复至室温,搅拌均匀,然后置于-80℃的冷浴中,冷却15min。加入0.35~0.5mlsec-buli(1.3m,溶剂为正己烷),保持-80℃反应15min。将干燥好的金刚烷取代的丙烯酸酯类单体cm4(1.5~2ml)的温度降至-60℃,滴入反应体系,保持-80℃反应40min。产物在乙醇中沉淀,得到白色固体3.5~4.5g。共聚单体三甲基硅取代苯乙烯(ptmss)、cm4及嵌段共聚物2的结构式分别如下:及实施例5嵌段共聚物6(pcm5-b-phfmba)的聚合以阴离子聚合为例,其制备步骤如下:将2~2.5ml金刚烷取代的丙烯酸酯类单体cm5和30~35ml正己烷用二丁基镁溶液(1m,溶剂为正己烷)在40℃下处理0.5h,转移至反应瓶中。将反应瓶恢复至室温,搅拌均匀,然后置于-80℃的冷浴中,冷却15min。加入0.35~0.5mlsec-buli(1.3m,溶剂为正己烷),保持-80℃反应15min。将干燥好的含氟丙烯酸酯类单体(1.5~2ml)的温度降至-60℃,滴入反应体系,保持-80℃反应40min。产物在乙醇中沉淀,得到白色固体2.5~4g。共聚单体含氟丙烯酸酯(phfmba)单体、cm5及嵌段共聚物6的结构式分别如下:及实施例6核磁共振谱图(1h-nmr)本发明使用400mhz傅里叶变换核磁共振波谱仪(avanceiii)测定了实施例1~5所得材料的具体结构,采用氘代氯仿、氘代四氢呋喃作为溶剂,通过对结构式中氢原子的特征出峰位置的峰积分,确定材料的结构、组分比例、聚合物分子量等信息。图1示出了实施例1所得材料的核磁共振谱图,嵌段ps和嵌段pcm1的特征h峰均与所示结构相对应,并且其积分面积也与两嵌段单体的投料比一致。图2示出了实施例2所得材料的核磁共振谱图,嵌段pmma和嵌段pcm2的特征h峰均与所示结构相对应,并且其积分面积也与两嵌段单体的投料比一致。图3示出了实施例3所得材料的核磁共振谱图,嵌段ppdfma和嵌段pcm3的特征h峰均与所示结构相对应,并且其积分面积也与两嵌段单体的投料比一致。图4示出了实施例4所得材料的核磁共振谱图,嵌段ptmss和嵌段pcm4的特征h峰均与所示结构相对应,并且其积分面积也与两嵌段单体的投料比一致。图5示出了实施例5所得材料的核磁共振谱图,嵌段phfmba和嵌段pcm5的特征h峰均与所示结构相对应,并且其积分面积也与两嵌段单体的投料比一致。实施例7凝胶渗透色谱(gpc)在本发明中,数均分子量(mn)和多分散性pdi可以通过凝胶色谱法测试(四氢呋喃相),使用通用校正方法校正即可,且以苯乙烯为校正基准,其测试结果如表1所示。表1凝胶渗透色谱测试结果嵌段共聚物mnpdi162001.14252001.24378001.11456001.136105001.20实施例8小角x射线散射(saxs)本发明使用小角x射线散射(saxs)测试聚合物材料组装的结构和尺寸,通过最高峰和次级峰的出峰位置以及比例计算其组装尺寸和微观形貌。检测过程如下:将干燥好的实施例1~5的嵌段共聚物溶解在四氢呋喃或甲苯中,随后滴涂在硅片上,在热板上加热,特定时间后取下,用冷板冷却。所得的样品用于后期测量saxs。由图6可知,嵌段共聚物ps-b-pcm1在退火后,其组装后得到full-pitch为12.6nm的结构,即half-pitch为6.3nm。由图7可知,嵌段共聚物pmma-b-pcm2在退火后,其组装后得到full-pitch为11.5nm的结构,即half-pitch为5.8nm。由图8可知,嵌段共聚物pcm3-b-ppdfma在退火后,其组装后得到full-pitch为14.6nm的结构,即half-pitch为7.3nm。由图9可知,嵌段共聚物ptmss-b-pcm4在退火后,其组装后得到full-pitch为10.7nm的结构,即half-pitch为5.4nm。由图10可知,嵌段共聚物pcm5-b-phfmba在退火后,其组装后得到full-pitch为17.4nm的结构,即half-pitch为8.7nm。实施例9刻蚀性能检测(rie)本发明使用反应离子刻蚀和膜厚仪来测试嵌段聚合物中两组分的抗刻蚀性能。例如嵌段共聚物由a和b两组分单体聚合而成,在测试刻蚀对比度时,需要先合成a单体均聚物以及b单体的均聚物,随后将其分别配置成均聚物物溶液旋涂在硅片基底上。采用膜厚仪测量其刻蚀前的原始膜厚(测量三次取平均值),随后对a和b两种均聚物的薄膜用反应离子刻蚀(采用cf4,o2,chf3,sf6,ar,h2,co2,n2等单一或多种气体混合),功率10w-500w,气体流速2-100sccm。刻蚀时间分别取5s,10s,15s,20s,30s,45s,60s……,并采用膜厚仪对不同刻蚀时间后的均聚物膜厚进行测量(三次取平均值)。对于a与b两种不同的均聚物薄膜,在相同刻蚀条件下膜厚减小的多少差异,做出其抗刻蚀对比度的计算和定义。对实施例1~5的嵌段共聚物进行测试,结果如下:ps-b-pcm1:在cf4等离子体刻蚀(此处以刻蚀气体cf4为例,气体流量为30sccm,功率为30w)下的两组分均聚物膜厚变化具有明显差异,其中,ps(原始膜厚为200nm)在1min的刻蚀后膜厚则减少了45nm,pcm1(原始膜厚为235nm)在1min的刻蚀后膜厚减少了26nm。可见,碳含量更高的金刚烷丙烯酸酯组分相较于苯乙烯组分的抗刻蚀性较强。pmma-b-pcm2:在氧气等离子体刻蚀(此处以刻蚀气体o2为例,气体流量为50sccm,功率为50w)下的两组分均聚物膜厚变化具有明显差异,其中,pmma(原始膜厚为280nm)在1min的刻蚀后膜厚则减少了90nm,pcm2(原始膜厚为250nm)在1min的刻蚀后膜厚减少了32nm。可见,碳含量更高的金刚烷丙烯酸酯组分相较于甲基丙烯酸酯组分的抗刻蚀性较强。pcm3-b-ppdfma:在氧气等离子体刻蚀(此处以刻蚀气体氧气为例,气体流量为50sccm,功率为30w)下的两组分均聚物膜厚变化具有明显差异,pcm3均聚物薄膜(原始膜厚为250nm)在1min的刻蚀后膜厚减少了28nm,ppdfma(原始膜厚为260nm)在1min的刻蚀后膜厚减少了90nm。可见,碳含量更高的金刚烷丙烯酸酯组分相较于含氟甲基丙烯酸酯组分的抗刻蚀性较强。ptmss-b-pcm4:在氧气等离子体刻蚀(此处以刻蚀气体氧气为例,气体流量为50sccm,功率为50w),其中,ptmss均聚物薄膜(原始膜厚为260nm)在1min的刻蚀后膜厚减少了20nm,pvm4(原始膜厚为230nm)在1min的刻蚀后膜厚减少了25nm。可见,金刚烷丙烯酸酯组分与硅取代的苯乙烯单体的抗刻蚀能力相近。pcm5-b-phfmba:在氧气等离子体刻蚀(此处以刻蚀气体氧气为例,气体流量为50sccm,功率为30w)下的两组分均聚物膜厚变化具有明显差异,其中,pcm5均聚物薄膜(原始膜厚为250nm)在1min的刻蚀后膜厚减少了30nm,phfmba(原始膜厚为240nm)在1min的刻蚀后膜厚减少了100nm。可见,碳含量更高的金刚烷丙烯酸酯组分相较于含氟甲基丙烯酸酯组分的抗刻蚀性较强。以上详细描述了本发明的具体实施例,应当理解,本领域的普通技术人员无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本
技术领域:
中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1