一种5-溴-2-甲基烟酸乙酯的合成方法与流程
本发明涉及有机合成
技术领域:
,具体涉及一种5-溴-2-甲基烟酸乙酯的合成方法。
背景技术:
:作为一种重要的医药和化工原料,5-溴-2-甲基烟酸乙酯广泛用于药物中间体的合成,其结构式如下:文献上关于其合成报道较少:2002年专利ep1180514从硝基丙二醛和氨基巴豆酸乙酯出发,经过三步反应合成了5-溴-2-甲基烟酸乙酯,但第一步的关环反应收率仅为30%,且硝基丙二醛为易爆品,储存和使用时安全风险高,因此这条合成路线不适合大规模工业化生产,其合成反应如下:2017年专利us2017/15655从丙烯醛出发,将其溴代后发生消去反应,制得2-溴丙烯醛,然后与2-氯代乙酰乙酸乙酯发生缩合反应,以7%的收率得到目标产物。2-溴丙烯醛极易发生聚合,且缩合反应收率低,杂质种类较多,纯化困难,难以进行放大生产,其合成反应如下:2000年meck公司开发了式i类似物5-氯-2-甲基烟酸乙酯的合成工艺,条件温和,可放大至百千克级别;但其工艺应用在5-溴-2-甲基烟酸乙酯(式i)的合成时,不能顺利得到产物,主要原因在于溴代中间体(式iii)的稳定性比氯代物(式iv)差,在强酸、强碱以及高温均容易发生分解,其合成反应如下:技术实现要素:本发明的目的在于提供一种5-溴-2-甲基烟酸乙酯的合成方法,以解决上述
背景技术:
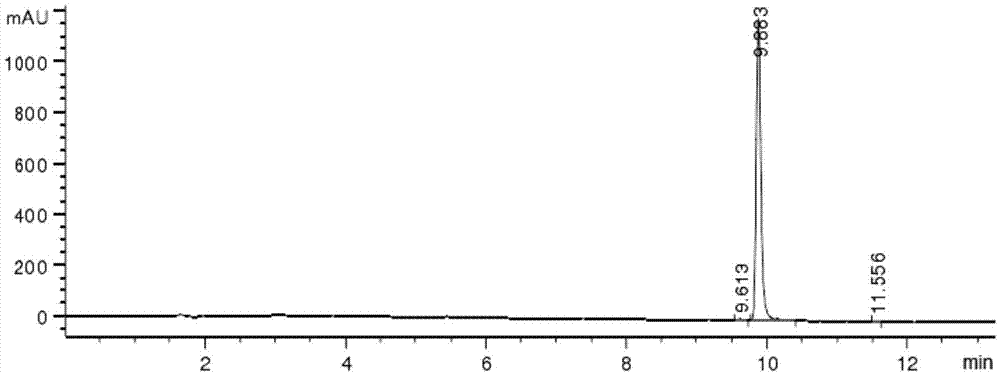
中提出的问题。为实现上述目的,本发明提供如下技术方案:一种5-溴-2-甲基烟酸乙酯的合成方法,其特征在于,包括以下步骤:1)将乙酰乙酸乙酯和碱a加入溶剂b中进行烯醇化;发生如下反应:2)向步骤(1)中加入中间体iii和dabco进行反应;发生如下反应:3)加入乙酸铵进行环合;发生如下反应:4)加入水淬灭,有机溶剂c萃取;5)除去有机溶剂c得待结晶粗品;6)将待结晶粗品重结晶得5-溴-2-甲基烟酸乙酯。作为本发明进一步的方案:步骤1)所述碱a为叔丁醇钾,所用质量为乙酰乙酸乙酯0.5-1.5倍。作为本发明进一步的方案:步骤1)所述的溶剂b为包括但不限于二氧六环、2-甲基-四氢呋喃、四氢呋喃、乙二醇二甲醚、甲基叔丁醚、乙醚、异丙醚中的一种醚类;溶剂b所用质量不大于乙酰乙酸乙酯质量的50倍。作为本发明进一步的方案:步骤2)所述中间体iii所用质量为乙酰乙酸乙酯质量3-4倍;dabco所用质量为乙酰乙酸乙酯质量0.5-1.0倍;反应温度为零下78-5℃,反应时间4-6h。作为本发明进一步的方案:步骤2)反应温度为零下10-0℃。作为本发明进一步的方案:步骤3)所用乙酸铵质量为乙酰乙酸乙酯质量1-2倍;反应温度为55-65℃,反应时间为15-17h。作为本发明进一步的方案:步骤4)所用水为质量为乙酰乙酸乙酯质量25-30倍的冰水;有机溶剂c为质量为乙酰乙酸乙酯质量6.5-7.5倍的mtbe。作为本发明进一步的方案:步骤6)所述重结晶温度为零下30-100℃。作为本发明进一步的方案:步骤6)所用重结晶溶剂为酯类、酮类、醚类、卤代烃、醇类、烷烃类中的一种或几种的混合物,所用重结晶溶剂质量不大于待结晶粗品质量的40倍。作为本发明进一步的方案:所述酯类包括但不限于乙酸乙酯和乙酸丙酯;所述酮类包括但不限于丙酮和丁酮;所述醚类包括但不限于二氧六环、2-甲基-四氢呋喃、四氢呋喃和乙二醇二甲醚;所述卤代烃包括但不限于二氯甲烷和氯苯;所述醇类包括但不限于甲醇、乙醇、正丙醇和异丙醇;所述烷烃类包括但不限于正戊烷、正己烷和正庚烷。与现有技术相比,本发明的有益效果是:以meck开发的5-氯-2-甲基烟酸乙酯合成工艺为基础,通过改变反应溶剂和降低反应温度,显著改善了中间体iii的稳定性,从而使得反应能顺利得到产物,后处理通过重结晶纯化,从而避免了耗时费力的柱层析工序,平均收率达到70%,纯度达到99.0%以上,适合用于大规模工业化生产。附图说明图1为实施例1合成的5-溴-2-甲基烟酸乙酯的液相色谱图;图2为对比例1合成的5-溴-2-甲基烟酸乙酯的核磁共振氢谱图;图3为对比例3合成的5-溴-2-甲基烟酸乙酯的液相色谱图。具体实施方式下面结合具体实施方式对本专利的技术方案作进一步详细地说明。实施例1250ml的三口烧瓶中加入乙酰乙酸乙酯(7.0克)和二氧六环(140克),冰盐浴下将体系温度降至-10℃,依次加入叔丁醇钾(6.4克)、中间体iii(23.8克)和dabco(6.1克),搅拌5小时,加入乙酸铵(10.0克),升温至60℃反应16小时,tlc监控至反应结束。反应液降至室温,倒入冰水(200克)中,加入mtbe(50克),搅拌1小时分层,水相mtbe(50克)提取一次,有机相加入无水硫酸钠(20克)干燥1小时。过滤,旋转蒸发仪浓缩,残留物用热的正庚烷(30.0克)溶解后,降温至-20℃,搅拌1小时,过滤,减压干燥得到5-溴-2-甲基烟酸乙酯,白色固体7.8克,收率60.0%,纯度99.4%(见表1)。其合成路线为:实施例2250ml的三口烧瓶中加入乙酰乙酸乙酯(7.0克)和2-甲基四氢呋喃(70克),冰盐浴下将体系温度降至-8℃,依次加入叔丁醇钾(3.5克)、中间体iii(21.0克)和dabco(3.5克),搅拌4小时,加入乙酸铵(7.0克),升温至55℃反应15小时,tlc监控至反应结束。反应液降至室温,倒入冰水(180克)中,加入mtbe(46克),搅拌1小时分层,水相mtbe(46克)提取一次,有机相加入无水硫酸钠(20克)干燥1小时。过滤,旋转蒸发仪浓缩,残留物用热的正庚烷(20.0克)溶解后,降温至0℃,搅拌1小时,过滤,减压干燥得到5-溴-2-甲基烟酸乙酯,白色固体6.4克,收率49.0%。实施例3250ml的三口烧瓶中加入乙酰乙酸乙酯(7.0克)和四氢呋喃(210克),冰盐浴下将体系温度降至-6℃,依次加入叔丁醇钾(7.0克)、中间体iii(22.5克)和dabco(4.5克),搅拌4小时,加入乙酸铵(9.0克),升温至57℃反应16小时,tlc监控至反应结束。反应液降至室温,倒入冰水(180克)中,加入mtbe(48克),搅拌1小时分层,水相mtbe(46克)提取一次,有机相加入无水硫酸钠(20克)干燥1小时。过滤,旋转蒸发仪浓缩,残留物用热的正庚烷(25.0克)溶解后,升至20℃,搅拌1小时,过滤,减压干燥得到5-溴-2-甲基烟酸乙酯,白色固体6.4克,收率49.0%。实施例4250ml的三口烧瓶中加入乙酰乙酸乙酯(7.0克)和二氧六环(280克),冰盐浴下将体系温度降至-4℃,依次加入叔丁醇钾(10.0克)、中间体iii(24.0克)和dabco(5.5克),搅拌5小时,加入乙酸铵(12克),升温至62℃反应16小时,tlc监控至反应结束。反应液降至室温,倒入冰水(205克)中,加入mtbe(51克),搅拌1小时分层,水相mtbe(51克)提取一次,有机相加入无水硫酸钠(20克)干燥1小时。过滤,旋转蒸发仪浓缩,残留物用热的正庚烷(35.0克)溶解后,升温至40℃,搅拌1小时,过滤,减压干燥得到5-溴-2-甲基烟酸乙酯,白色固体6.5克,收率50.1%。实施例5250ml的三口烧瓶中加入乙酰乙酸乙酯(7.0克)和四氢呋喃(340克),冰盐浴下将体系温度降至-2℃,依次加入叔丁醇钾(10.5克)、中间体iii(28.0克)和dabco(7.0克),搅拌5小时,加入乙酸铵(14.0克),升温至65℃反应17小时,tlc监控至反应结束。反应液降至室温,倒入冰水(210克)中,加入mtbe(52克),搅拌1小时分层,水相mtbe(52克)提取一次,有机相加入无水硫酸钠(20克)干燥1小时。过滤,旋转蒸发仪浓缩,残留物用热的正庚烷(40.0克)溶解后,升温至80℃,搅拌1小时,过滤,减压干燥得到5-溴-2-甲基烟酸乙酯,白色固体5.6克,收率43.2%。对比例1参考专利us2017/15655:1l的三口烧瓶中加入丙烯醛(3.6克),加入水(100毫升),搅拌溶解后,降温至0℃,慢慢滴加溴素(10.6克),滴完后保温1小时,加入二氯甲烷萃取(50毫升)。合并有机相,依次用饱和氯化钠水溶液(10毫升)以及水洗涤,无水硫酸钠(5克)干燥有机相。过滤,旋转蒸发仪浓缩,得到2-溴-丙烯醛(6.0克),该产品为淡黄色液体,在0℃放置时不稳定,逐渐会变为泡沫状固体。因此得到的2-溴-丙烯醛不进行纯化,直接进行下一步反应。室温下将2-溴-丙烯醛(6.0克)、乙酸铵(71.8克)、2-氯代乙酸乙酯(25.7克)溶解在乙酸(40毫升)中,室温下搅拌48小时,tlc显示原料消失。加入冰水(400毫升)淬灭反应,水相用乙酸乙酯(100毫升*3次)萃取。合并有机相,依次用饱和氯化钠水溶液(10毫升)以及水洗涤,无水硫酸钠(5克)干燥有机相。过滤,旋转蒸发仪浓缩,得到粗品,快速柱层析纯化,淋洗剂:乙酸乙酯/石油醚=1:8,得到白色固体2.8克,收率7.2%。其合成路线为:对比例2参考meck公司工艺,制备氯代物:250ml的三口烧瓶中加入乙酰乙酸乙酯(7.0克)和四氢呋喃(140毫升),冰盐浴下将体系温度降至-10℃。加入叔丁醇钾(6.4克),升温至20℃后依次加入化合物iv(20.7克)和1,4-二叠氮双环[2,2,2]辛烷(6.1克),搅拌1小时后升温至45℃,继续搅拌3小时,加入乙酸铵(10.0克),升温至60℃反应16小时,tlc监控至反应结束。反应液降至室温,倒入冰水(200毫升)中,加入mtbe(50毫升),搅拌1小时分层,水相mtbe(50毫升)提取一次,有机相加入无水硫酸钠(20克)干燥1小时。过滤,旋转蒸发仪浓缩,残留物用正庚烷(50.0克)重结晶,得到氯代物v,白色固体7.0克,收率65.0%,其合成路线为:对比例3参考meck公司工艺,制备溴代物:250ml的三口烧瓶中加入乙酰乙酸乙酯(7.0克)和四氢呋喃(140毫升),冰盐浴下将体系温度降至-10℃。加入叔丁醇钾(6.4克),升温至20℃后依次加入化合物iv(20.7克)和1,4-二叠氮双环[2,2,2]辛烷(6.1克),搅拌1小时后升温至45℃,继续搅拌3小时,加入乙酸铵(10.0克),升温至60℃反应16小时,tlc监控至反应结束。反应液降至室温,倒入冰水(200毫升)中,加入mtbe(50毫升),搅拌1小时分层,水相mtbe(50毫升)提取一次,有机相加入无水硫酸钠(20克)干燥1小时。过滤,旋转蒸发仪浓缩,残留物用正庚烷(50.0克)重结晶,得到氯代物v,白色固体1.2克,收率9.1%,纯度98.5%(见表2),其合成路线为:表1实施例1合成的5-溴-2-甲基烟酸乙酯的纯度表表2对比例3合成的5-溴-2-甲基烟酸乙酯的纯度表表3不同合成方法合成5-溴-2-甲基烟酸乙酯的收率表项目产物收率实施例1溴代物60.0%实施例2溴代物49.0%实施例3溴代物49.0%实施例4溴代物50.1%实施例5溴代物43.2%对比例1溴代物7.2%对比例2氯代物65.0%对比例3溴代物9.1%从表3中可以看出本发明实施例1-5的合成工艺得到的产品收率在40.0%以上,明显高于目前方法对比例1和对比例3的收率,本发明纯化工艺简单,得到产品纯度高。对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1