合成索非布韦关键中间体的方法与流程
本发明涉及药物合成领域,具体而言,涉及一种合成索非布韦关键中间体的方法。
背景技术:
索非布韦是首个无需联合干扰素就能安全有效治疗慢性丙肝的药物。在其合成过程中,n4-苯甲酰-2’-c-甲基-β-d-阿拉伯呋喃基胞苷是其关键中间体。
现有技术中有多种合成该关键中间体的方法,例如:以胞嘧啶核苷为起始原料合成该关键中间体,合成路线如下式所示:
上述路线采用选择性苯甲酰化、醇羟基保护、氧化、加成、脱保护、保护反应将胞苷氨基转化为尿苷羰基。该反应中使用1,3-二氯-1,1,3,3-四异丙基二硅氧烷对糖苷上的羟基进行保护,完成甲基取代后使用tbaf进行脱保护得到sf-2。该步脱保护收率仅为61%,对整体收率有较大影响。
目前,大多数合成方法主要也都是采用1,3-二氯-1,1,3,3-四异丙基二硅氧烷对糖苷上的羟基进行保护,完成甲基取代后再使用tbaf进行脱保护。该方法脱保护收率通常在40~70%之间,处于低收率水平,且生产成本高,对环境有一定的污染。
有鉴于此,特提出本发明。
技术实现要素:
本发明的第一目的在于提供一种合成索非布韦关键中间体的方法,所述方法反应效率高,副产物少、收率高,降低后处理难度,减少对环境的污染,分离纯化操作简便。
为了实现本发明的上述目的,特采用以下技术方案:
一种合成索非布韦关键中间体的方法,包括以下步骤:
a.在微波反应器中加入醇和原料化合物,所述原料化合物的结构式为:
b.体系缓慢升温至第一温度,开启所述微波反应器并调节功率至第一功率进行反应;
c.反应结束后浓缩,然后降温至第二温度,搅拌、抽滤得到所述索非布韦关键中间体,所述索非布韦关键中间体的结构式为:
采用微波水解的方法,摒弃传统的碱性脱保护反应体系,采用微波水解手段,使得反应物分子在高频电场作用下,从原有的随机分布状态转向依照电场的极性排列取向,而高频电场的频率不断发生变化,使得其排列取向随之发生变化,在这一过程中造成分子的运动并发生相互摩擦,使得介质温度升高。基于此,缩短反应时间,提高反应收率,降低生产成本及对环境的污染,得到高质量的索非布韦关键中间体n4-苯甲酰-2’-c-甲基-β-d-阿拉伯呋喃基胞苷。
优选地,所述步骤a中还加入乙酸。
在反应体系中加入乙酸,有助于原料在相对温和的条件下反应,提升反应效率和收率。
优选地,所述醇与所述原料化合物的体积重量比为1.5-2.0ml:1g。
对原料的使用量的限定,有利于进一步提高反应收率。
优选地,所述醇为甲醇、乙醇或异丙醇,优选甲醇。
对醇的优选,主要是为了提供合适的质子溶剂,提高反应效率和收率。
优选地,所述第一温度为50-90℃。
更加优选地,所述第一温度为80℃。
对温度的优选,一方面是为了控制反应的剧烈程度和副反应的发生,减少副产物,提高收率;另一方面也是为了达到收率和反应效率的平衡,实现高效快速反应的同时,保证高收率。
优选地,所述第一功率为400-700w。
更加优选地,所述第一功率为600w。
对微波反应器的功率的控制,实际上也是为了控制反应进程,避免反应进行过快或过慢,导致高效率低收率或高收率低效率现象。
优选地,所述第二温度为0-5℃。
对于降温终点的控制,是为了保证产品的收率和纯度。
可选地,反应时间为30-45分钟。
反应时间的优选,是为了控制反应进程,快速、高效、高收率得到产品。
与现有技术相比,本发明的有益效果为:
(1)采用微波水解方式进行脱保护反应,反应效率高,副产物少,产品收率高、纯度高;
(2)后处理难度低,分离纯化操作简便;
(3)对环境的污染少。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,以下将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
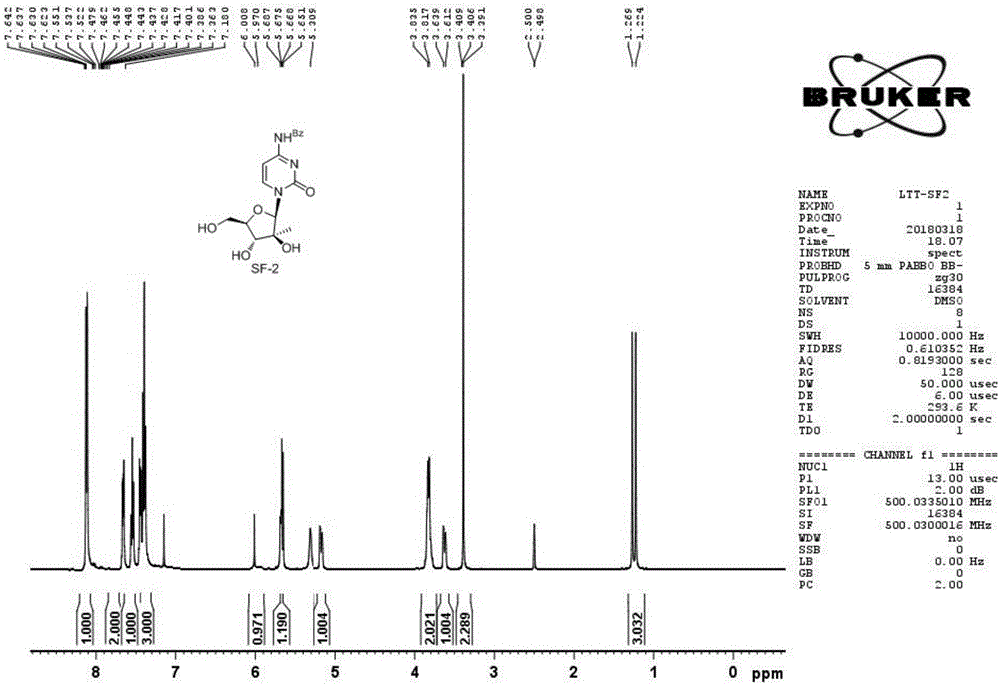
图1为实施例1提供的产品的h谱图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
实施例1
在微波反应器中加入50ml甲醇,开启搅拌,加入原料化合物30g,缓慢升温至50℃,开启微波反应器(型号xh-mc-1)调节设备功率在700w,反应时间30分钟,停止反应,浓缩后降温至5℃搅拌30分钟,抽滤得到索非布韦关键中间体的固体,收率89%,纯度99.1%。
原料化合物的结构式为:
索非布韦关键中间体为n4-苯甲酰-2’-c-甲基-β-d-阿拉伯呋喃基胞苷,结构式为:
实施例2
在微波反应器中加入45ml乙醇,开启搅拌,加入原料化合物30g和乙酸2g,缓慢升温至90℃,开启微波反应器(型号xh-mc-1)调节设备功率在400w,反应时间45分钟,停止反应,浓缩后降温至0℃搅拌30分钟,抽滤得到索非布韦关键中间体的固体,收率88%,纯度99.0%。
实施例3
在微波反应器中加入50ml甲醇,开启搅拌,加入原料化合物30g和乙酸2g,缓慢升温至80℃,开启微波反应器(型号xh-mc-1)调节设备功率在600w,反应时间40分钟,停止反应,浓缩后降温至3℃搅拌30分钟,抽滤得到索非布韦关键中间体的固体,收率92%,纯度99.2%。
实施例4
在微波反应器中加入60ml异丙醇,开启搅拌,加入原料化合物30g和乙酸2g,缓慢升温至70℃,开启微波反应器(型号xh-mc-1)调节设备功率在500w,反应时间35分钟,停止反应,浓缩后降温至2℃搅拌30分钟,抽滤得到索非布韦关键中间体的固体,收率90%,纯度99.1%。
取实施例1得到的产品结构鉴定,核磁检测谱图如图1所示。对实施例2-4得到产品进行结构鉴定,与实施例1相同。
1hnmr(dmso-d6)数据解析如下:δ1.20(s,3h),3.62-3.69(m,2h),3.73-3.78(m,2h),5.19(t,1h,j=5.4hz),5.25(s,1h),5.52(d,1h,j=5.0hz),5.99(s,1h),7.32(d,1h,j=5.8hz),7.50(t,2h,j=7.7hz),7.62(t,1h,j=7.3hz),8.00(d,2h,j=7.3hz),8.14(d,1h,j=6.9hz),11.22(s,1h);13cnmr(dmso-d6)δ19.6,61.3,77.5,78.5,85.2,88.9,95.2,128.5,132.8,133.3,147.3,154.9,162.9,167.4。
比较例
按照现有技术中的反应方法进行对照试验:
将与本申请相同的所述原料化合物加入thf中,然后依次加入乙酸和四丁基氟化铵,在室温下反应,反应结束后用硅胶处理并真空浓缩干燥,得到的棕褐色残留固体用硅胶柱层析处理,得到所述索非布韦关键中间体,得率为61%。
与现有的tbaf(四丁基氟化铵)反应体系相比较,区别如下表1所示:
表1本申请与现有tbaf反应体系区别
需要特别说明的是,虽然现有反应体系的纯度比本申请的纯度高0.2%,但是现有方法反应结束后需要使用柱层析纯化,成本高、效率低、收率进一步大幅下降。
本申请提供的合成索非布韦关键中间体的方法,采用微波水解方式进行脱保护反应,脱去双tips(三异丙基硅基)保护基团,反应效率大大提高,仅需要30-45分钟即可完成整个反应;此外,显著提高反应收率,减少反应的副产物,采用微波水解使得反应收率提高至88-92%;无需进行色谱柱纯化处理,降低生产工艺后处理难度,减少可能对环境造成的污染,后处理过程分离纯化操作简便。
尽管已用具体实施例来说明和描述了本发明,然而应意识到,在不背离本发明的精神和范围的情况下可以作出许多其它的更改和修改。因此,这意味着在所附权利要求中包括属于本发明范围内的所有这些变化和修改。
- 还没有人留言评论。精彩留言会获得点赞!