使用肿瘤教育的血小板的针对癌症的群智能增强的诊断和治疗选择的制作方法
本发明属于医学诊断领域,特别是疾病诊断和监测领域。本发明涉及用于检测疾病的标志物、用于检测疾病的方法,以及用于确定疾病治疗的疗效的方法。
背景技术:
癌症是发达国家中的主要死亡原因之一。研究表明,许多癌症患者在更难以治疗的晚期被诊断出来。癌症主要由正常细胞中的连续突变而驱动,导致dna损伤并最终导致明显的基因改变而带来癌变状态。
癌症通常基于肿瘤标志物来诊断。肿瘤标志物是存在于癌细胞中或在响应癌症的另一种细胞中所产生的物质。一些肿瘤标志物也存在于正常细胞中,但是在癌变细胞中例如以更高水平的替代形式存在。通常可以在液体样品(诸如血液、尿液、粪便或体液)中鉴定肿瘤标志物。
目前大多数使用的肿瘤标志物是蛋白质。一个实例是前列腺特异性抗原(psa),其用作前列腺癌的肿瘤标志物。大多数单个肿瘤标志物对于患有癌症的个体患者的管理是不可靠的。替代性标志物,例如,基因表达水平和dna改变(诸如dna甲基化),已经开始用作肿瘤标志物。鉴定多个基因的表达水平和/或基因组dna的改变可以改善癌症的检测、诊断、预后和治疗。需要广泛的数据挖掘和统计分析来发现能够区分正常变异与癌变状态的肿瘤标志物的组合。
基于血液的液体活检,包括经肿瘤教育的血小板(tumor-educatedbloodplatelets)(teps;nilssonetal.,2011.blood118:3680-3683;bestetal.,2015.cancercell28:666-676;nilssonetal.,2015.oncotarget7:1066-1075)已成为癌症的非侵入性检测和治疗选择的有前景的生物标志物来源。公知的挑战在于从这种液体生物源中鉴定最佳生物标志物组。为了选择用于疾病分类的稳健生物标志物组,提出了“群智能(swarmintelligence)”的使用,尤其是粒子群优化(particleswarmoptimization,pso)(kennedyetal.,2001.themorgankaufmannseriesinevolutionarycomputation.ed:davidb.fogel;bonyadiandmichalewicz2016.evolutionarycomputation:1-54;kennedyandeberhart,1995.proceedingsofieeeinternationalconferenceonneuralnetworks:1942-1948)。
pso驱动的算法受到相伴的鸟群和鱼群的启发,它们通过自组织有效地适应其环境或鉴定食物来源。生物信息学上,pso算法被用于鉴定复杂参数选择程序的最佳解决方案,包括生物标志物基因列表的选择(alshamlanetal.,2015.computationalbiolchem56:49-60;martinezetal.,2010.computationalbiolchem34:244-250)。
技术实现要素:
靶向治疗和个性化医疗在很大程度上取决于疾病分析和伴随诊断的发展。疾病来源的核酸中的突变对靶向治疗的响应可以是高度预测性的。然而,获得易得到的高质量核酸仍然是一个重要的发展障碍。血液通常每微升含有150000-350000个凝血细胞(thrombocytes)(血小板),为研究和临床应用提供了高度可用的生物标志物来源。此外,凝血细胞分离相对简单,并是血库/血液学实验室的标准程序。由于凝血细胞不含有细胞核,因此它们功能维持所需的rna转录来源于凝血细胞起源过程中的骨髓巨核细胞。另外,凝血细胞可以在循环期间通过各种转移机制从其他细胞中摄取rna和/或dna。例如,肿瘤细胞释放大量遗传物质的收集,其中一些由突变rna形式的微泡分泌。在血液循环期间,凝血细胞可以吸收癌细胞和其他患病细胞分泌的遗传物质,作为癌症伴随诊断的有吸引力的平台,特别是在个性化医疗的背景下。
本发明提供了一种对癌症患者施用免疫疗法的方法,所述免疫疗法调节程序性死亡蛋白1(pd-1)与其配体之间的相互作用,所述方法包括提供来自患者样品的步骤,所述样品包含从所述患者的无核细胞获得的mrna产物;确定所述样品中表1中列出的至少4个基因、更优选表1中列出的至少5个基因、更优选表1中列出的至少6个基因的基因表达水平;比较经确定的基因表达水平与参考样品中所述基因的参考表达水平;根据与参考文献的比较,将患者归为所述免疫治疗的阳性响应者,或归为非阳性响应者;并且对被归为阳性响应者的癌症患者施用免疫疗法。
在本发明的优选方法中,确定表1中列出的至少4个基因、更优选表1中列出的至少5个基因、更优选表1中列出的至少6个基因、更优选表1中列出的至少10个基因、更优选表1中列出的至少50个基因、更优选表1中列出的所有基因的基因表达水平。
调节pd-1与其配体pd-l1或pd-l2之间相互作用的所述免疫疗法旨在激活免疫系统以攻击患者的癌症。抑制pd-1与其配体之间相互作用的已知调节剂包括单克隆抗体(如atezolizumab(genentechoncology/roche)、avelumab(merck/pfizer)、durvalumab(astrazeneca/medimmune)、nivolumab(bristol-myerssquibb)、lambrolizumab(merck)、pidilizumab(curetech)和pembrolizumab单抗(merck)),以及融合蛋白(如amp-224(glaxosmithkline))。优选的免疫疗法包括nivolumab。
在另一个实施方案中,本发明提供了一种对受试者样品存在或不存在肺癌进行归类的方法,包括提供来自受试者的样品的步骤,其中样品包含从所述受试者的无核细胞获得的mrna产物;确定表2中列出的至少5个基因的基因表达水平;比较经确定的基因表达水平与参考样品中所述基因的参考表达水平;基于经确定的基因表达水平与所述参考基因表达水平之间的比较,将所述样品归类为存在或不存在肺癌。
所述受试者,哺乳动物,优选人,不知道患有肺癌。所述肺癌优选是非小细胞肺癌。
在本发明的优选方法中,确定表2中列出的至少10个基因,更优选表2中列出的至少45个基因,更优选表2中列出的至少50个基因,更优选表2中列出的所有基因的基因表达水平。
如上所述,无核细胞可在肿瘤发生和癌症转移期间充当局部和全身响应者,从而暴露于肿瘤介导的教育(tumor-mediatededucation),并导致行为改变。无核细胞(诸如凝血细胞)可以作为rna生物标志物发现物来检测和分类来自不同来源的癌症。存在于无核细胞中的所述rna优选源自肿瘤细胞,并从肿瘤细胞转移至无核细胞。这些无核细胞可以容易地从液体活检(诸如血液)中分离,并且可以含有来自有核肿瘤细胞的rna。
包含mrna产物的所述样品优选从液体活检,优选血液获得。所述无核细胞优选为或包含凝血细胞。在一个优选的实施方案中,从血液样品中分离凝血细胞,并随后从所述经分离的凝血细胞中分离mrna。
所述样品中表1中列出的至少4个基因,更优选表1中列出的至少5个基因,和/或表2中列出的至少5个基因的基因表达水平,可以通过本领域已知的任何方法测定,这样的方法包括基于微阵列的分析、基因表达的系列分析(sage)、多重聚合酶链式反应(pcr)、多重连接依赖性探针扩增(mlpa)、基于珠子的多路复用(诸如luminex/xmap),以及包括下一代测序的高通量测序。基因表达水平优选通过下一代测序确定。
本发明进一步提供一种通过向患者分配调节pd-1与其配体之间的相互作用的免疫疗法,治疗癌症患者、优选肺癌患者的方法,其中,通过归类来自患者的样品而选择所述癌症患者,所述样品包含从所述受试者的无核细胞获得的mrna产物;确定表1中列出的至少4个基因,更优选表1中列出的至少5个基因的基因表达水平;比较经确定的基因表达水平与参考样品中所述基因的表达水平;基于与参考的比较,将患者归入所述免疫治疗的阳性响应者,或作为非阳性响应者;并将免疫疗法分配给被选为阳性响应者的癌症患者。
进一步提供了调节pd-1与其配体之间的相互作用的免疫疗法,用于治疗癌症患者、优选肺癌患者的方法中,其中,通过归类来自患者的样品来选择所述癌症患者,所述样品包括从所述受试者的无核细胞获得的mrna产物;确定表1中列出的至少4个基因,更优选表1中列出的至少5个基因的基因表达水平;比较经确定的基因表达水平与参考样品中所述基因的表达水平;基于与参考的比较,将患者归入所述免疫治疗的阳性响应者,或作为非阳性响应者;并将免疫疗法分配给被选为阳性响应者的癌症患者。
如上所述,调节pd-1与其配体pd-l1或pd-l2之间相互作用的所述免疫疗法旨在活化免疫系统以攻击患者的癌症。抑制pd-1与其配体之间相互作用的已知调节剂包括单克隆抗体(如atezolizumab(genentechoncology/roche)、avelumab(merck/pfizer)、durvalumab(astrazeneca/medimmune)、nivolumab(bristol-myerssquibb)、lambrolizumab(merck)、pidilizumab(curetech)和pembrolizumab单抗(merck)),以及融合蛋白(诸如amp-224(glaxosmithkline))。优选的免疫疗法包括nivolumab。
本发明进一步提供了获得生物标志物组的方法,所述生物标志物组用于归类来自受试者的样品,该方法包括从具有情况a的受试者的液体样品中分离无核细胞、优选凝血细胞。从经分离的细胞中分离rna;确定所述经分离的rna中至少100个基因的rna表达水平;确定来自不具有情况a的受试者的对照样品中所述至少100个基因的rna表达水平;以及使用基于粒子群优化的算法来获得区分具有情况a的受试者与不具有情况a的受试者的生物标志物组。
优选地,具有情况a的受试者患有癌症、优选肺癌,或者对癌症治疗具有已知的阳性响应,而没有情况a的受试者未患有癌症,或者对癌症治疗具有已知的阴性响应。
附图说明
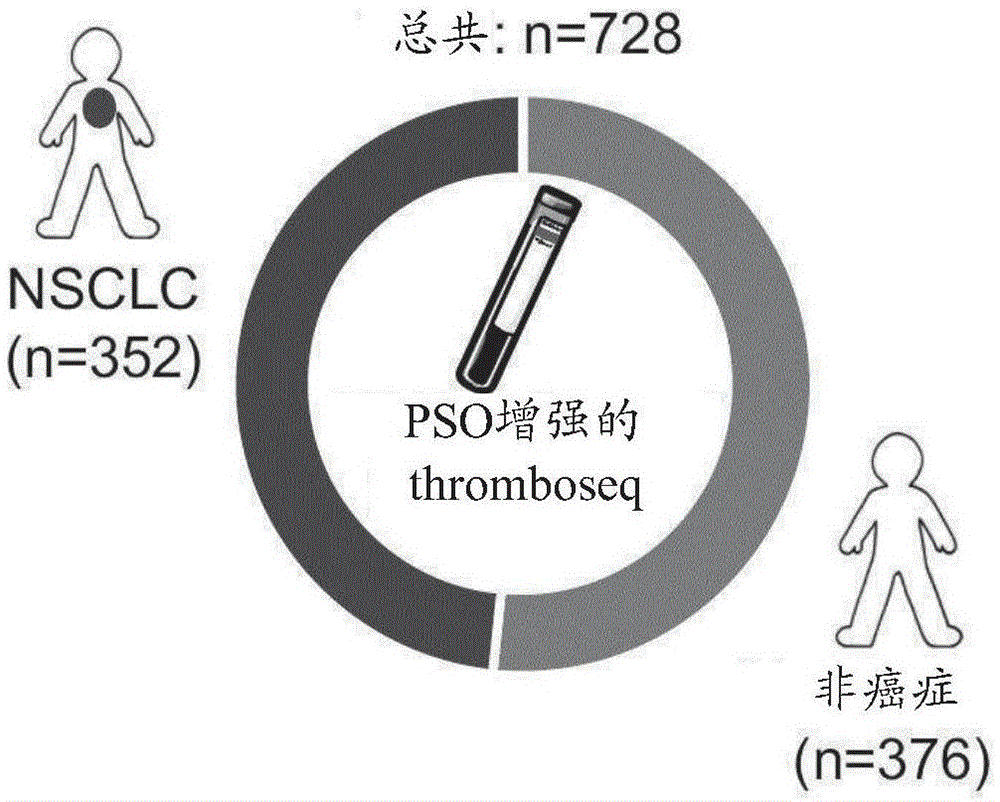
图1.用于nsclc诊断的pso增强的thromboseq
(a)针对thromboseq的该研究中包括的非癌症和nsclc血小板样品(总共728个)的概述。(b)可变剪接分析概述,对tep特征的估计贡献以及与这些分析相关的其他图。rbp=rna结合蛋白(c)粒子群智能方法的示意图。浅灰色到深灰色的点代表使用thromboseq分类算法,使用100个随机选择的参数(左)或100个通过群智能选择的参数(右)分类的38个样品的auc值。出于可视化目的,将点镜像(mirrored)两次。用星号在两个图中都显示了通过群增强的thromboseq达到的最佳auc值。(d)使用与癌症年龄及血液储存时间匹配的非癌症和nsclc组群的群增强的thromboseq分类的roc分析。灰色虚线表示通过loocv评估的训练群组的roc评估,红线表示评估群组的roc评估(n=40),蓝线表示验证群组的roc评估(n=130)。表示的是群组大小、最佳准确度和auc值。acc.=准确度。(e)在roc曲线中总结的完整728个样品群组中评估的群增强的thromboseq算法的性能。群智能利用评估群组(红线,n=88个样品)通过选择生物标志物基因小组来优化120个样品训练群组的分类性能。使用患者年龄和/或血液储存时间不匹配的群组(n=520,蓝线)验证群增强的thromboseqnsclc诊断算法。通过loocv评估的训练群组的性能用灰色虚线表示。表示的是群组大小、最佳准确度和auc值。acc.=准确度。
图2-基于tep的nivolumab响应预测
(a)实验装置的示意图。在治疗开始前一个月(基线,t=0)包括了符合pd-1抑制剂nivolumab治疗条件的患者的血液。基于ct成像并根据recist1.1标准读出的肿瘤响应在nivolumab治疗开始后的6-8周、3个月和6个月进行。选择最佳响应作为总体肿瘤响应(参见实施例1)。(b)响应者(蓝色,n=44)和无响应者(红色,n=60)的群智能驱动的基因小组选择后无监督的血小板mrna聚类的热图。(c)104个nivolumab基线样品的群体thromboseqnivolumab响应预测算法的roc分析。通过loocv方法测量的训练群组性能由红线表示,依赖评估群组由黑线表示,独立验证群组由蓝线表示。灰色实线(上限)和点线(下限)线表示由随机训练的算法产生的roc曲线。黑点表示用于最佳治疗选择和非响应者排除的算法的潜在临床阈值。(d)2x2交叉表,表明独立验证群体的分类准确性,其中thromboseq分类读数针对排除值进行了优化。100%的灵敏度导致53%的特异性。表示的是样品编号和百分比。
图3-实验方法thromboseq
(a)用于癌症诊断和治疗监测的基于thromboseq机器学习的液体活检的示意图。从具有不同疾病的个体和健康个体的血小板产生的rna-seq文库用作thromboseq算法开发的输入。在使用群模块和模型验证进行算法优化之后,该平台实现了基于rna特征的疾病分类和治疗监测。(b)训练、评估和验证群组的示意图和样品群组详细信息。群组用于评估群增强的thromboseq的分析性能,并用于在患者年龄及血液储存时间匹配群组中研究诊断分类能力。患者年龄及血液储存时间匹配的群组在130个样品的训练群组中得到验证,使用40个样品的评估群组进行优化。
图4-thromboseq的技术性能参数
(a)患者年龄及血液储存时间匹配的血小板样品群组(n=263)的人口统计特征概述。显示了非癌症(n=104)和nsclc(n=159)个体的特征。表示的每个临床组是男性个体的数量和总数的百分比、中位年龄(包括四分位范围(iqr)和最小和最大年龄,以年为单位)、吸烟状况和总数百分比,以及原发性nsclc向其他器官的转移(是/否)。n.a.=不可用。(b)通过流式细胞术分析从健康供体收集并使用thromboseq血小板分离方案分离的n=3(8小时时间点)或n=6(其他时间点)血小板样品测量血小板活化标志物的概述。浅灰色和深灰色框表示在表面上分别表达p-选择素或cd-63的血小板的平均百分比。框表示四分位距(iqr),黑线表示中位数,虚线表示1.5xiqr。点表示用trap活化血小板后这些表面标志物的表达(参见实施例1)。使用thromboseq血小板分离方案血小板样品仅进行最低限度的活化。(c)从在edta包被的vacutainer管中6ml全血中分离的以纳克为单位的每微升的血小板总rna产量的总结。通过bioanalyzerrnapicochip分析测量rna浓度和质量。对于非癌症(n=86)和nsclc(n=151),分别在箱形图中总结总rna产量。框表示四分位差(iqr),黑线表示中位数,虚线表示1.5xiqr。与非癌症患者相比,nsclc患者的血小板具有显著更高的rna产量(p=0.0014,双侧独立学生t检验)。(d)使用thromboseq方案的smartercdna合成和扩增的线性和效率。估计的rna输入(x轴,以pg/μl为单位)与输出smartercdna产量(y轴,以nm为单位,总共n=177次观察)的相关图。每个点代表一个样品,由临床组进行颜色编码。通过bioanalyzerpicochiprna测量的~500pg的平均rna输入用于smartercdna合成和pcr扩增。rna输入和cdna输出成正相关(r=0.23,p=0.003,pearson相关性)。(e)使用thromboseq方案的truseqcdna文库制备和pcr扩增的线性和效率。将smartercdna产量用作输入(x轴,以nm为单位)与输出的truseq血小板cdna序列文库产量(y轴,以nm为单位,总共n=177次观察)的相关图。每个点代表一个样品,由临床组进行颜色编码。除用于生物分析仪分析的1.5μl纯化缓冲液等分试样外,所有smartercdna输出均用作truseq文库制备的输入。smartercdna产量和truseq血小板cdna文库输出成正相关(r=0.44,p<0.0001,pearson相关性)。(f)具有突起的、平滑的和中间级突起的/平滑的剖面的样品的生物分析仪痕迹。对于每个实施例,显示了picochipbioanalyzer上的总rna,dna高灵敏度生物分析仪上的smarter扩增cdna和dna7500生物分析仪上的truseqcdna文库。x轴表示产物的长度(rna的核苷酸(nt)和cdna的碱基对(bp)),而y轴表示通过bioanalyzer2100测量的相对荧光。从突起到平滑的smartercdna样品,观察到smartercdna生物分析仪斜率的平滑度逐渐增加,而总rna和truseqcdna显示出无法区分的特征。(g)由smarter扩增产生的以nm为单位的相对cdna产量(上图),突起的、平滑的和中间级突起的/平滑的smartercdna组的以bp为单位的相对cdna长度(中图),以及内含子剪接rna读数的数量(下图)的概况。通过bioanalyzercdnahighsensitivity芯片上的图下面积测量cdna浓度。cdna产量在三种不同的smarter谱中是相当的。通过在bioanalyzer软件中选择200-9000bp区域来测量相对cdna长度。smartercdna斜率与平均cdna长度密切相关。映射到基因间区域的读数的贡献确实对符合thromboseq分析的跨越内含子的读数的数量产生负面影响。每个smarter斜率和临床组的样品数量显示在图表下方。框表示四分位差(iqr),黑线表示中位数,虚线表示1.5xiqr。(h)针对突起的(上)和平滑的(下)样品(每个n=50,随机取样)映射到基因间区域的读数的平均片段长度的直方图。将映射到基因间区域的重叠读数合并(参见在线方法),并对所得的总片段大小进行定量。突起的和平滑的样品主要含有<250nt的碎片,峰值在100-200nt区域。(i)选择用于thromboseq分析的内含子剪接rna读数。叠加图表示从跨越内含子的、外显子、内含子、基因间和线粒体区域亚指定(subspecified)每个样品的读数分布。值得注意的是,从映射到外显子区域的读数中减去跨越内含子的读数。根据跨越内含子的读数的比例(y轴)对样品(n=263)进行分类。(j)选择具有>3000个基因的样品用于thromboseq分析。图表显示对于经历thromboseq的740个血小板rna样品,跨越内含子的读数的总数(x轴)和检测到的基因数(y轴),具有至少一个跨越内含子的读数。检测到的基因数量与每个样品产生的跨越内含子的读数的总数部分相关。检测到的基因少于3000个(n=10)的样品被排除在分析之外。(k)使用浅度thromboseq(平均10-20百万读数)在血小板rna样品中以置信度(即>30个剪接rna读数)检测到的基因数量的总结,显示非癌症(n=377)和nsclc(n=353)群组。框表示四分位距(iqr),黑线表示中位数,虚线表示1.5xiqr。每个样品的基因的平均检测是约4500种不同的rna,并且与非癌症个体相比,在nsclc患者的血小板中平均略微降低。(1)浅度thromboseq与深度thromboseq的比较。从健康对照中收集的总共12个血小板rna样品进行深度thromboseq(每个样品的中位数59.7(最小-最大:43.2-96.2)百万个原始读数计数),并与匹配的浅度thromboseqrna-seq数据进行比较。对于深度thromboseq,从血小板总rna开始重新制备具有相当的输入浓度的血小板样品用于测序。图表显示了通过所有样品的中位读数计数(x轴)分类的每个基因的原始读数计数(对数转换的y轴)。突出显示在深度thromboseq中具有最高表达的三个基因。(m)留一样品法的互相关。为了研究一个样品(测试案例)与所有其他样品(参考群组)的可比性,我们进行了互相关,在此期间每个样品的计数与所有其他样品的中位计数相关。在选择检测到具有足够数量的基因的样品之后,将该步骤作为质量控制步骤(参见在线方法)被包括(也参见(j))。计算730次互相关,即将所有样品从参考群组中排除一次。结果表明,所有样品都显示出高样品间pearson相关性。样品间相关性<0.5(n=2)的样品被排除在分析之外。
图5-neclc患者的tep中的差异化剪接rna
(a)非癌症(n=104)和nsclc(n=159)个体之间差异化剪接rna的无监督层次聚类。总共1625个基因(698上升,927下降)表现出显著性,错误发现率<0.01(参见实施例3)。列表示样品,行表示基因,颜色强度表示z分数转换的rna表达值(在进行基于ruv的迭代校正模块的可视化之前)。样品聚类显示非随机分区(p<0.0001,费希尔精确检验)。(b)pagoda基因本体分析(参见实施例1)。对显著富集的基因进行无偏基因簇鉴定和基因本体分析。通过调整的z分数(表明高统计学显著性)的最显著结果被聚类和可视化。格雷码表示每个基因簇的每个样品的暗到亮(从低到高)得分。与非癌症样品相比,nsclc样品中的剪接评分较低的最显著的生物组(最大调整z分数为13.9)包括与翻译、rna结合蛋白(rbp)和信号传导相关的基因本体。与非癌症个体相比,nsclc患者中最显著富集的基因簇与信号传导和免疫应答相关(最大调整的z分数为5.3)。该聚类分析鉴定了非癌症个体血小板中血小板稳态基因特征与nsclc患者tep中特异性免疫信号传导途径之间的相关性。rbp=rna结合蛋白。
图6-thrombo剪接
(a)示意图代表读数分布分析方法。从患者年龄及血液储存时间匹配的群组中,我们将100bp读数映射到人类基因组并定量了映射到四个不同区域的读数(参见实施例3),即外显子、内含子和基因间区域(一起为‘基因组区域’)和线粒体基因组(缩写为mtdna)的数量。值得注意的是,跨越内含子的剪接的读数包括在外显子区域中。(b)箱形图显示非癌症(浅灰色,n=104)和nsclc(深灰色,n=159)映射到线粒体(mtdna)、外显子、内含子或基因间区域的读数的中位数和扩散,以及跨越内含子的和外显子边界读数的中位数和扩散。框表示四分位距(iqr),黑线表示中位数,虚线表示1.5xiqr。跨越内含子的读数定义为从外显子a开始并在外显子b结束的读数。外显子边界读数定义为覆盖相邻外显子-内含子边界的读数。将外显子、内含子、基因间、跨越内含子的和外显子边界读数标准化为一百万个总基因组读数。(c)替代rna异构体分析的总结图。示意图代表异构体计数矩阵的发展。为此,将92bp修整的rna-seq读数映射到人类基因组,并随后进行miso算法。miso算法使得能够从单个读取rna-seq数据推断已表达的rna异构体。将miso输出数据解卷积为计数矩阵,其包含每个表达的rna异构体的每个样品支持该特定异构体的读数的数量。将104个非癌症个体和159个nsclc患者的计数矩阵用于差异化表达分析。选择显著性值(fdr)<0.01的异构体。每个基因的差异化剪接rna异构体总数(fdr<0.01,n=743,总结在颜色代码中)的饼图(n=571,总结在饼图的饼中),表明非癌症和nsclc每个亲本基因之间显著改变的异构体的分布。在38%的显著改变的rna异构体中,多种异构体属于相同的亲本基因,支持一些基因显示多种rna异构体的共同调节的概念。基因总数(总共n=571)的饼图显示所有rna异构体共同增加的表达水平(277/571,49%)、共同降低的表达水平(281/571,49%)或替代拼接(13/571,2%)。实施例2中提供了其他详细内容。(d)外显子跳跃事件分析的总结图。示意图表示用于检测外显子跳跃事件的实验方法。使用miso算法映射和分析读数,其推断有利于特定外显子的包含(在示意图的顶部)或排除(在示意图的底部)的读数。为此,该算法还将考虑映射到相邻外显子的读数。在对大多数样品群组中的平均读取覆盖率进行筛选后(参见在线方法),总共230个外显子仍然有资格进行分析。由miso输出的百分比拼接(psi)值用于差异anova统计。在非癌症或nsclc样品中共鉴定出27个外显子可能被跳过(fdr<0.01)。直方图显示了psi值的方向,其中阳性psi值有利于在非癌症中排除,而阴性psi值有利于在nsclc中排除。经注释事件的基因名称(按fdr值分类并针对唯一基因名称进行筛选)列在框中。实施例2中提供了其他详细内容。
图7-p选择素特征
(a)映射到外显子坐标(x轴)的读数与p-选择素的对数转换的、ruv校正的和每百万计数的比例的相关图。每个点代表由临床组编码的样品(nsclc,n=159,深灰色,和非癌症,n=104,浅灰色)。外显子读数与p-选择素的表达水平相关(r=0.51,p<0.001)。(b)4722个基因的对数转换的每百万计数水平与p-选择素的对数转换的每百万计数之间的相关系数的分布。基因的子集显示与p-选择素的强相关性(r近似为-1或1),而其他基因则不显示(r近似为0)。对于直方图,使用0.05的箱尺寸。(c)在nsclctep特征中上调的基因(698个基因,也参见图5a),以及与p-选择素(selp特征,1820个基因)具有显著正相关(fdr<0.01)的基因的维恩图重叠。在tep特征中增加的基因的77%(536/698)也存在于selp特征中,表明selp特征可能部分地促成tep特征。
图8-tep衍生的rna特征的rna结合蛋白(rbp)分析
(a)示意性生物模型,突出了在翻译调节的背景下有核细胞和无核血小板之间的差异。有核细胞(左)能够通过转录因子(tf)介导的dna转录调节和维持转录组,从而引起蛋白质翻译。无核血小板缺乏基因组dna,因此缺乏通过tf调节rna含量的能力。循环血小板保留选择性剪接前mrna库的能力,表明在诱导剪接事件期间的关键调节功能。(b)rbp-thrombo搜索引擎算法的示意图。该算法被设计用于鉴定基因组的特定基因组区域中rbp基序序列的存在之间的相关性,这里应用于5′-utr和3′-utr。首先,该算法从人类基因组中提取感兴趣区域的参考序列(hg19)。此外,该算法补充了先前鉴定的经验证的rbp结合位点基序序列(rayetal.,2013.nature499:172-177)。通过减少基序序列,将547个非冗余寡核苷酸序列与utr参考序列匹配,并且将所有匹配计数(范围0至460)总结在utr至基序矩阵中,用于下游分析。有关rbp-thrombo搜索引擎算法的进一步详细信息,参见实施例1。(c)utr读数覆盖率筛选器。对该分析中包括的utr区域(n=19180,x轴)定量映射读数的数量(y轴)。具有超过五个(5′-utr)或三个(3′-utr)映射读数的utr被认为存在于血小板中。蓝点代表所有样品的平均计数,灰色阴影表示各自的标准偏差。(d)每个utr区域富集经鉴定的rbp结合位点。x轴和y轴代表每个rbp5′-utr和3′-utr的平均结合位点(点,n=102)。几种rbp在3′-utr中特异性富集,而其他rbp在5′-utr中富集(也参见实施例4)。(e和f)在血小板中检测到足够覆盖率的所有rbp(n=80,行)和所有5′-utr(e)和3′-utr(f)(对于5′-utr,n=3210,以及对于3′utr,n=3720,列,参见实施例4)区域的热图。结合位点的数量由热图颜色反映(参见灰度)。rbp的utr调节似乎是由rbp结合位点的存在/不存在介导的。(g)在nsclc/非癌差异化剪接分析中n个rbp结合位点与基因的对数倍数变化(logfc)之间的相关性分析(n=4722)(也参见图5a)。正相关表明结合位点的富集随logfc的增加,而负相关表明相反。图表示spearman的相关系数(x轴)与为多重假设检验(fdr)调整的伴随p值之间的关系。结果表明rbp停靠位点涉及nsclc和非癌症之间的基因的logfc。
图9-pso增强的thromboseq分类算法的示意图,以及与患者年龄及血液储存时间相匹配的nsclc和非癌症群组的应用。
(a)在thromboseq中实施的迭代校正模块的示意图。rna-seq数据校正程序包括多个步骤,即1)筛选低丰度基因、2)确定混杂变量中的稳定基因、3)原始读取计数去除基于不需要变量(ruv)的因子分析和校正,以及4)参考组介导的每百万计数和tmm标准化(也参见实施例1)。详细地,在步骤1中,排除了具有检测低置信度的基因,即超过90%的样品群组中少于30个跨越内含子的的剪接rna读数。在示意性实例中,两个上部基因(行)包含>90%的样品(在该示意性实施例中总共n=10)足够数量的读数,如浅灰色框所示。因此,将包括这些基因用于分析。较低的两个框表示具有足够数量基因的样品数量不足,因此促使算法从下游分析中去除这些特定基因。其次,该算法搜索在所有其他样品中显示稳定表达模式的基因。为此,该算法在(潜在的混杂)变量和原始读取计数之间进行多个pearson相关性分析,从而生成相关系数的分布。在示意图中,显示了跨越内含子的读数库大小(左)和患者年龄(右)。相关分布如下所示,推定的阈值(也经过pso选择,参见(e))用黑线表示。值得注意的是,随着原始跨越内含子的读数计数通过之后的每百万计数标准化,稳定基因必须近似相关系数为1(参见图9b-c)。在第三步中,算法首先使用ruvseq校正模块(ruvg函数)以无偏的方式鉴定对数据有贡献的因子。ruvseq校正方法基于基因子集的广义线性模型和通过奇值分解来估计和校正感兴趣的协变量和不想要的变量的贡献。其次,该算法迭代地将感兴趣的变量(组)和潜在的混淆变量(患者年龄及血液储存时间)与由ruvseq鉴定的因子相关联。如果确定一个因子与混杂因子相关(例如,“因子1”中的跨越内含子的读取文库大小),该因子将被标志物为去除(“移除”)。或者,如果确定一个因子与感兴趣的因子相关(例如,“因子2”中的组)或者没有一个因子被确定为相关因子(例如“因子3”),则该因子将不被删除(′保持′)。最后,在第四步骤中,仅使用来自训练群组的样品作为合格样品来进行默认的每百万计数标准化和m值的修正平均值(tmm)校正,以计算tmm校正因子。(b)相同的跨越内含子的文库大小的相同例子,如a.2(左)所示,但这里y轴表示每百万计数(cpm)标准化计数。该图强调,对于该特定变量,必须选择高达1的相关系数,引起在cpm标准化后选择稳定的基因。(c)cpm标准化后所有基因的四分位数范围分布通过与文库大小的相关性排序。与相关系数减小的样品(黑线的左侧)相比,高度相关的基因(黑线右侧,示例性阈值r>0.8)显示cpm标准化后的最小四分位数范围。(d)使用我们先前的方法(上图)和新方法(当前研究,下图)标准化的263个样品的相对对数表达(rle)图。rle图表示读取计数与样品中位计数的对数比,并且应该为良好标准化的数据集显示以零为中心的类似分布。校正模块显著降低了样品间的差异性(p<0.0001,双侧学生t检验)。(e)群增强的thromboseq分类模块的示意图。该算法的多个步骤和筛选器被群优化,如“鸟”标志所示。首先,数据集经历迭代校正模块(参见图9a)。其次,计算和选择大多数差异化剪接的基因(参见实施例1)。第三,去除在第二步中选择的基因之间高度相关的基因。第四,使用训练群组建立svm模型,通过网格搜索优化伽马(g)和成本(c)参数(参见在线方法)。第五,根据对svm模型的贡献递归地对选择用于分类的所有基因进行排序,得到排序的分类基因列表。该列表经历基于群的筛选。第六,使用简化基因列入更新的svm模型,再次通过网格搜索进行伽马(g)和成本(c)优化。第七,通过第二粒子群优化算法(参见在线方法)进一步优化伽马(g)和成本(c)值。最后,使用简化基因列表和经优化的伽马(g)和成本(c)参数,构建最终的svm模型。
图10-在nivolumab治疗开始后2-4周,nsclc患者的teprna谱的比较分析。(a)n=17个响应者和n=11个非响应者的差异化剪接分析,其在开始治疗后2-4周收集血液。195个基因小组显示响应者和非响应者之间的显著分离(通过群智能优化的基因小组,通过fisher精确检验p<0.0001)。维恩图显示1246个基因的基线响应预测特征和195个基因的基线随访响应预测特征具有最小重叠。(b)n=61个响应者和n=72个非响应者的差异化剪接分析,其中在基线和治疗开始后2-4周内收集血液。(c)在治疗的响应者的tep中鉴定了378种改变的rna,且在治疗的非响应者的tep中鉴定了107种改变的rna(通过群智能优化的基因组,通过fisher精确检验p<0.0001)。维恩图显示两个特征都具有最小的叠加。
具体实施方式
(1)缩写
如本文所用,术语“癌症”是指由致癌转化细胞的增殖导致的疾病或紊乱。“癌症”应被视为包括多种良性或恶性肿瘤中的任何一种或更多种,包括能够通过人体或动物体或其部分(诸如通过淋巴系统和/或血流)侵入性生长和转移的肿瘤。如本文所用,术语“肿瘤”包括良性和恶性肿瘤或固体生长物,尽管本发明特别涉及恶性肿瘤和实体癌的诊断或检测。癌症进一步包括但不限于癌(carcinomas)、淋巴瘤或肉瘤(诸如卵巢癌、结肠癌、乳腺癌、胰腺癌、肺癌、前列腺癌、泌尿道癌、子宫癌、急性淋巴性白血病、霍奇金病、肺小细胞癌、黑色素瘤、神经母细胞瘤、神经胶质瘤(如胶质母细胞瘤)、软组织肉瘤、淋巴瘤、黑色素瘤、肉瘤和腺癌)。在本发明方面的优选实施方案中,放弃凝血细胞癌症。
如本文所用,术语“液体活检”是指从受试者获得的液体样品。所述液体活检优选选自血液、尿液、乳汁、脑脊髓液、间质液、淋巴液、羊水、胆汁、耳垢、粪便、雌性射出的液体(femaleejaculate)、胃液、粘液心包液、胸膜液、脓液、唾液、精液、包皮垢、痰、滑液、汗液、眼泪、阴道分泌物和呕吐物。优选的液体活检是血液。
如本文所用,术语“血液”是指全血(包括血浆和细胞)并且包括动脉血、毛细血管血和静脉血。
如本文所用,术语“无核血细胞”是指缺乏细胞核的细胞。该术语包括红细胞和凝血细胞。根据本发明的无核细胞的优选实施方案是凝血细胞。术语“无核血细胞”优选不包括由于细胞分裂错误而缺乏细胞核的细胞。
如本文所用,术语“凝血细胞”是指血液血小板,即小的、不规则形状的细胞碎片,其不具有含dna的细胞核并且在哺乳动物的血液中循环。凝血细胞直径为2-3μm,并且源自前体巨核细胞的分片。尽管它们保留了一些巨核细胞衍生的mrna作为其直系起源的一部分,血小板或凝血细胞缺乏核dna。凝血细胞的平均寿命为5至9天。凝血细胞参与并在止血中起重要作用,引起血栓形成。
(2)确定基因表达水平
本发明描述了基于分析无核细胞(诸如从血液中提取的凝血细胞)中的基因表达水平来诊断、预言或预测对治疗的响应的方法。这种方法稳健且容易。这归因于快速和直接的提取程序和提取的核酸的质量。在临床环境中,从血液样品提取凝血细胞在一般的生物样品采集中实施,并因此可以预见到临床的实施相对容易。
本发明提供了使用所述一般方法诊断、预言或预测对治疗的响应的一般方法。当在本文中提及本发明的方法时,除非另外明确指出,否则提及任何和所有这些实施方案。
本发明的方法可以在包含无核血细胞的任何合适的身体样品(诸如包含血液的组织样品)上进行,但优选所述样品是全血。
受试者的血液样品可以通过任何标准方法(如通过静脉提取)而获得。
所需的血液量不受限制。取决于所采用的方法,技术人员将能够确定进行本发明方法的各个步骤所需的样品量并获得足够的核酸用于遗传分析。通常,这样的量将包括0.01μl至100ml、优选1μl至10ml、更优选约1ml的体积。
可在收集样品后立即分析体液,优选血液样品。或者,根据本发明的方法的分析可以在储存的体液或其无核细胞(优选凝血细胞)的储存部分上进行。可以使用本领域已知的方法和装置保存用于测试的体液或其无核血细胞的部分。在无核血细胞部分中,凝血细胞优选保持在灭活状态(即处于非活化状态)。以这种方式,细胞完整性和疾病衍生的核酸被最佳地保存。来自体液的含凝血细胞的样品优选不包括血小板贫乏的血浆或血小板富含的血浆(prp)。为了获得最佳分辨率,优选进一步分离血小板。
体液,优选血液样品可以适当地被加工,例如,可以被纯化或消化,或者可以从中提取特定化合物。可以通过本领域技术人员已知的方法从样品中提取无核细胞,并将其转移到任何合适的培养基中以提取核酸。可以处理受试者的体液以去除核酸降解酶如rna酶和dna酶,以防止核酸的破坏。
从受试者的身体样品中提取凝血细胞可涉及任何可用的方法。在输血医学中,凝血细胞通常通过单采血液成分术收集,其是一种医疗技术,其中供体或患者的血液通过分离出一种特定成分的装置并将其余部分返回循环。用专门的离心机分离各个血液成分。单采血小板术(也称为血小板提取术或血小板单采)是收集凝血细胞的单采血液成分术。现代自动单采血小板术使得献血者能够给出他们的一部分凝血细胞,同时保留他们的红细胞和至少一部分血浆。尽管可以通过单采血液成分术提供如本文所设想的包含凝血细胞的体液,但通常更容易收集全血并通过离心从中分离出凝血细胞细胞部分。通常,在这样的方案中,首先通过在室温下约20分钟的约120×g的离心步骤将凝血细胞与其他血细胞分离,以获得富含血小板的血浆(prp)部分。然后洗涤凝血细胞,例如在磷酸盐缓冲盐水/乙二胺四乙酸中洗涤,以除去血浆蛋白并富集凝血细胞。洗涤步骤通常在室温下以850-1000×g离心约10分钟。可以进行进一步的富集以产生更纯的凝血细胞部分。
血小板分离通常涉及在含有抗凝剂柠檬酸盐右旋糖的vacutainer管(例如36ml柠檬酸、5mmol/lkcl、90mmol/lnacl、5mmol/l葡萄糖、10mmol/ledta,ph6.8)中收集血液样品。ferretti等人描述了用于血小板分离的合适方案(ferrettietal..2002.jclinendocrinolmetab87:2180-2184)。该方法包括初步离心步骤(每10分钟1300rpm)以获得血小板富含的血浆(prp)。然后可以在抗聚集缓冲液(tris-hcl10mmol/l;nacl150mmol/l;edta1mmol/l;葡萄糖5mmol/l;ph7.4)中洗涤血小板三次并如上所述离心,以避免任何血浆蛋白污染和去除任何残留的红细胞。然后可以在4000rpm下进行20分钟的最后离心以分离血小板。对于定量确定,血小板膜的蛋白质浓度可用作内参。可以使用血清白蛋白作为标准,通过bradford(bradford,1976.analbiochem72:248-254)的方法确定这种蛋白质浓度。
包含无核细胞的样品可以在收获时新鲜制备,或者可以制备并在-70℃下储存直至用于样品制备的处理。优选地,在保持无核细胞的核酸含量的质量的条件下进行储存。防腐的条件的实例是使用例如福尔马林固定和石蜡包埋、添加rnase抑制剂(诸如rnasin(pharmingen)或rnasecure(ambion))、添加水性溶液(诸如rnalater(assuragen;us06204375)、介导有机溶剂保护效果的hepes-谷氨酸缓冲液(hope;de10021390)和rcl2(alphelys;wo04083369),以及添加非水性溶液(诸如通用分子固定件(sakurafinetekusainc.;us7138226))。
确定基因表达水平的方法是技术人员已知的,并且包括但不限于northern印迹、定量pcr、微阵列分析和rna测序。优选同时确定所述基因表达水平。可以例如通过多重qpcr、rna测序程序和微阵列分析进行同时分析。微阵列分析使得能够同时确定大量基因表达(诸如超过50个基因、超过100种基因、超过1000种基因、超过10000种基因,甚至基于全基因组)的基因表达水平,使得能够在本发明的方法中,使用大量基因表达数据来标准化所确定的基因表达水平。
基于微阵列的分析涉及使用固定在固体表面(阵列)上的所选生物分子。微阵列通常包含核酸分子,称为探针,其能够与基因表达产物杂交。将探针暴露于已标记的样品核酸,杂交,并确定样品中与探针互补的基因表达产物的丰度。微阵列上的探针可包含dna序列、rna序列或dna和rna的共聚物序列。探针还可以包含dna和/或rna类似物(诸如核苷酸类似物或肽核酸分子(pna),或其组合)。探针的序列可以是基因组dna的完整或部分片段。序列也可以是体外合成的核苷酸序列(诸如合成的寡核苷酸序列)。
探针优选对表1-3中列出的基因的基因表达产物具有特异性。当探针包含与基因表达产物或其cdna产物的核苷酸序列完全互补的连续核苷酸区段时,探针是特异性的。当探针包含与所述基因的基因表达产物或其cdna产物的核苷酸序列部分互补的连续核苷酸区段时,探针也可以是特异性的。部分地表示来自至少20个核苷酸的连续区段中的核苷酸的最多5%不同于所述基因的基因表达产物的相应核苷酸序列。术语互补在本领域中是已知的,并且是指通过碱基配对规则与待检测的序列相关的序列。优选仔细设计探针序列以最小化与所述探针的非特异性杂交。优选探针是单链核酸分子或模拟单链核酸分子。所述互补连续核苷酸区段的长度可在15个碱基和几千个碱基之间变化,优选在20个碱基和1000个碱基之间,更优选在40和100个碱基之间,最优选约60个核苷酸。最优选的探针包含约与基因或其cdna产物的基因表达产物的核苷酸序列相同的60个核苷酸。在本发明的方法中,可以使用包含如表1-3和5-7中所示的探针序列的探针。
为了通过微阵列确定基因表达水平,优选直接或间接标记样品中的基因表达产物,并在有利于在探针和已标记的基因表达产物样品中的互补分子之间形成双链体的条件下与阵列上的探针接触。可以确定在洗涤微阵列后保持与探针相关的标记的量,并将其用作与所述探针互补的核酸分子的基因表达水平的测量。
用于确定基因表达水平的优选方法是通过测序技术,优选rna样品的下一代测序(ngs)技术。已经开发了用于测序rna的测序技术。这种测序技术包括,例如,合成测序。合成测序或循环测序可以通过逐步添加含有例如可切割或可光漂白的染料标志物的核苷酸来完成,例如,如美国专利no.7,427,673;美国专利no.7,414,116;wo04/018497;wo91/06678;wo07/123744;和美国专利no.7,057,026中所描述的。或者,可以使用焦磷酸测序技术。随着特定核苷酸掺入新生链中,焦磷酸测序检测到无机焦磷酸(ppi)的释放(ronaghietal.,1996,analyticalbiochemistry242:84-89;ronaghi,2001.genomeres11:3-11;ronaghietal.,1998.science281:363;美国专利no.6,210,891;美国专利no.6,258,568;和美国专利no.6,274,320)。在焦磷酸测序中,可以检测到释放的ppi,因为它通过atp硫酸化酶立即转化为三磷酸腺苷(atp),并通过荧光素酶产生的光子检测产生的atp水平。
测序技术还包括通过连接技术测序。这些技术使用dna连接酶掺入寡核苷酸并鉴定这些寡核苷酸的掺入,尤其在美国专利no.6,969,488;美国专利no.6,172,218;和美国专利no.6,306,597中描述。其他测序技术包括例如原位荧光测序(fisseq)和大规模并行标志物测序(mpss)。
测序技术可以通过直接测序rna,或通过测序rna至cdna转化的核酸文库来进行。用于测序rna样品的大多数方案采用在测序之前将样品中的rna转化为双链cdna形式的样品制备方法。
经确定的基因表达水平优选进行标准化。标准化是指用于调整或校正用于确定基因表达水平的测量中系统误差的方法。系统偏差可能是由于总体性能差异的变化、无核细胞分离效率的差异导致分离的无核细胞纯度的差异,以及由于例如纯度的变化的rrna样品之间的差异。在确定基因表达水平期间,可以在处理样品期间引入系统偏差。
(3)确定的基因表达水平的比较
将样品中表1-3的确定的基因表达水平与参考样品中相同基因的表达水平进行比较。所述比较可以生成索引分数(indexscore),其指示个体、受试者或患者的样品中已确定的表达水平与参考样品中的表达水平的相似性。例如,可以通过确定来自已被归类为患有癌症的个体获得的样品的基因表达的中位数值与来自被归类为未患癌症的个体获得的样品的基因表达的中位数值之间的倍数变化/比率来生成索引。可以例如在anova(方差分析)模型中检验这种倍数变化/比率在两个分别的组之间显著的相关性。可以在模型中计算单变量p值,并且经过多次校正检验(benjamini&hochberg,1995.jrss,b,57:289-300)可以用作确定不同组之间基因表达显示明显差异的显著性的阈值。还可以在将协变量(诸如肿瘤阶段/等级/大小)添加到anova模型中的情况下进行多变量分析。
类似地,可以通过患者样品中基因的表达水平与已知对调节pd-1和其配体之间的相互作用的免疫疗法有响应的一个或更多个癌症样品中表达水平的平均值或平均值(mean)之间的pearson相关系数,和与已知对调节pd-1和其配体之间的相互作用的免疫疗法无响应的一个或更多个癌症样品中表达水平的平均值或平均值(mean)之间的pearson相关系数来确定索引。得到的pearson分数可用于提供索引分数。所述分数可以在表示完美的相似性的+1和表示反向相似性的-1之间变化。优选地,使用任意阈值来将样品归类为响应或不响应。更优选地,基于相应的最高相似性测量,将样品分类为响应的或不响应的。优选地,相似性得分被显示或输出到用户接口设备、计算机可读存储介质或本地或远程计算机系统。
为了预测调节pd-1与其配体之间相互作用的免疫疗法的响应,所述参考样品优选包含从已知对所述免疫疗法做出阳性响应的个体的无核细胞和/或已知不对所述免疫疗法做出阳性响应的个体的无核细胞获得的基因表达产物。类似地,对于存在或不存在癌症的受试者样品的归类,所述参考样品优选包含从已知患有癌症和/或已知不患有癌症的个体的无核细胞获得的基因表达产物。
所述参考样品优选地提供至少2个独立个体,更优选至少5个独立个体,更优选至少10个独立个体(诸如10-100个个体)的无核细胞中基因表达的平均或平均水平的测量。
参考样品的无核细胞中所述基因的平均或平均表达水平优选呈现在用户界面装置、计算机可读存储介质或本地或远程计算机系统上。存储介质可以包括但不限于软盘、光盘、只读光盘存储器(cd-rom),可重写光盘(cd-rw)、记忆棒和磁光盘。
(4)预测对调节pd-1与其配体之间相互作用的免疫疗法的施用的响应
表1中列出的至少4个基因,更优选表1中列出的至少5个基因的基因表达水平可用于在施用所述治疗之前,预测对调节pd-1及其配体的相互作用的免疫疗法对癌症患者的响应。
为此,从已知患有癌症(诸如肺癌)的患者中分离出无核细胞、优选凝血细胞。从分离的无核细胞中分离包含核糖核酸(rna)、优选信使rna(mrna)的样品。在使用本领域技术人员已知的任何方法将rna逆转录成脱氧核糖核酸(cdna)拷贝后,标记所得的cdna并且例如通过下一代测序,例如在illumina测序平台上定量基因表达水平。
基于测序结果,在包含来自所述癌症患者的核糖核酸(rna)的样品中确定表1中列出的至少4个基因的基因表达水平,更优选表1中列出的至少5个基因,并且优选进行标准化。将标准化的表达水平与参考样品中的表1中列出的相同的至少4个基因,更优选至少5个基因的表达水平进行比较。所述参考样品从一个或更多个已知对调节pd-1与其配体之间的相互作用的免疫疗法的阳性响应的癌症患者获得,和/或从一个或更多个已知对调节pd-1与其配体之间的相互作用的免疫疗法的阴性响应的癌症患者获得。从所述比较中,获得了预测的对施用调节pd-1与其配体之间的相互作用的免疫疗法(诸如施用nivolumab)的响应效力。
本文考虑的是对已知患有癌症、尤其是肺癌的受试者的样品进行归类的方法,包括提供来自受试者样品的步骤,其中样品包含从所述受试者的无核细胞获得的mrna产物;确定表1中列出的至少4个基因,更优选表1中列出的至少5个基因的基因表达水平;将所述已确定的基因表达水平与参考样品中所述基因的参考表达水平进行比较;并且基于已确定的基因表达水平与参考基因表达水平之间的比较,对所述样品进行归类以评估对调节pd-1与其配体之间的相互作用的免疫疗法(诸如施用nivolumab)响应的可能性。
在根据本发明的优选方法中,确定表1中列出的至少4个基因,更优选表1中的至少5个基因的表达水平,更优选表1中至少10个基因的表达水平,更优选表1中至少20个基因的表达水平,更优选表1中至少30个基因的表达水平,更优选表1中至少40个基因的表达水平,更优选表1中至少50个基因的表达水平,更优选表1中所有532个基因的rna表达水平。
进一步优选的是,来自表1的至少5个基因包含表1中列出的前4个基因,更优选如表1中所示的具有最低p值的前5个基因,更优选如表1中所示的具有最低的p值的前10个基因,更优选如表1所示的具有最低p值的前20个基因,更优选如表1所示的具有最低p值的前30个基因,更优选如在表1中所示的具有最低p值的前40个基因,更优选如表1中所示的具有最低p值的前50个基因。
在进一步优选的实施方案中,所述在表1中列出的至少4个基因,更优选来自表1至少5个基因包括ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)和ensg00000126698(dnajc8);更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)和ensg00000121879(pik3ca);更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)和ensg00000174238(pitpna);更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)、ensg00000174238(pitpna)和ensg00000084754(hadha);更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)、ensg00000174238(pitpna)、ensg00000084754(hadha)和ensg00000272369);更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)、ensg00000174238(pitpna)、ensg00000084754(hadha)、ensg00000272369)和ensg00000073111(mcm2);更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)、ensg00000174238(pitpna)、ensg00000084754(hadha)、ensg00000272369)、ensg00000073111(mcm2)、ensg00000137073(ubap2)、ensg00000115866(dars)、ensg00000229474(patl2)、ensg00000086589(rbm22)、ensg00000145675(pik3r1)、ensg00000088833(nsfl1c)、ensg00000267243、ensg00000260661、ensg00000144747(tmf1)和ensg00000158578(alas2)、更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)、ensg00000174238(pitpna)、ensg00000084754(hadha)、ensg00000272369)、ensg00000073111(mcm2)、ensg00000137073(ubap2)、ensg00000115866(dars)、ensg00000229474(patl2)、ensg00000086589(rbm22)、ensg00000145675(pik3r1)、ensg00000088833(nsfl1c)、ensg00000267243、ensg00000260661、ensg00000144747(tmf1)、ensg00000158578(alas2)、ensg00000083642(pds5b)、ensg00000142089(ifitm3)、ensg00000107175(creb3)、ensg00000162585(c1orf86)、ensg00000142687(kiaa0319l)、ensg00000100796(smek1)、ensg00000142856(itgb3bp)、ensg00000103479(rbl2)、ensg00000048471(snx29)、ensg00000196233(lcor)和ensg00000068120(coasy):更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)、ensg00000174238(pitpna)、ensg00000084754(hadha)、ensg00000272369)、ensg00000073111(mcm2)、ensg00000137073(ubap2)、ensg00000115866(dars)、ensg00000229474(patl2)、ensg00000086589(rbm22)、ensg00000145675(pik3r1)、ensg00000088833(nsfl1c)、ensg00000267243、ensg00000260661、ensg00000144747(tmf1)、ensg00000158578(alas2)、ensg00000083642(pds5b)、ensg00000142089(ifitm3)、ensg00000107175(creb3)、ensg00000162585(c1orf86)、ensg00000142687(kiaa0319l)、ensg00000100796(smek1)、ensg00000142856(itgb3bp)、ensg00000103479(rbl2)、ensg00000048471(snx29)、ensg00000196233(lcor)、ensg00000068120(coasy)、ensg00000120868(apaf1)、ensg00000198265(helz)、ensg00000162688(agl)、ensg00000228215、ensg00000147457(chmp7)、ensg00000129187(dctd)、ensg00000141644(mbd1)、ensg00000172172(mrpl13)、ensg00000110697(pitpnm1)和ensg00000102054(rbbp7):更优选ensg00000084234(aplp2)、ensg00000165071(tmem71)、ensg00000143515(atp8b2)、ensg00000119314(ptbp3)、ensg00000126698(dnajc8)、ensg00000121879(pik3ca)、ensg00000174238(pitpna)、ensg00000084754(hadha)、ensg00000272369)、ensg00000073111(mcm2)、ensg00000137073(ubap2)、ensg00000115866(dars)、ensg00000229474(patl2)、ensg00000086589(rbm22)、ensg00000145675(pik3r1)、ensg00000088833(nsfl1c)、ensg00000267243、ensg00000260661、ensg00000144747(tmf1)、ensg00000158578(alas2)、ensg00000083642(pds5b)、ensg00000142089(ifitm3)、ensg00000107175(creb3)、ensg00000162585(c1orf86)、ensg00000142687(kiaa0319l)、ensg00000100796(smek1)、ensg00000142856(itgb3bp)、ensg00000103479(rbl2)、ensg00000048471(snx29)、ensg00000196233(lcor)、ensg00000068120(coasy)、ensg00000120868(apaf1)、ensg00000198265(helz)、ensg00000162688(agl)、ensg00000228215、ensg00000147457(chmp7)、ensg00000129187(dctd)、ensg00000141644(mbd1)、ensg00000172172(mrpl13)、ensg00000110697(pitpnm1)、ensg00000102054(rbbp7)、ensg00000153214(tmem87b)、ensg00000150054(mpp7)、ensg00000122008(polk)、ensg00000151150(ank3)、ensg00000165970(slc6a5)、ensg00000100811(yy1)、ensg00000152127(mgat5)、ensg00000172493(aff1)、ensg00000213722(ddah2)、ensg00000177425(pawr)、ensg00000260017、ensg00000141429(galnt1)、ensg00000119979(fam45a)、ensg00000136167(lcp1)、ensg00000244734(hbb)、ensg00000143569(ubap2l)、ensg00000079459(fdft1)、ensg00000197459(hist1h2bh)和ensg00000080371(rab21)。
在最优选的实施方案中,来自表1的一组至少4个基因包含ensg00000164985(psip1)、ensg00000114316(usp4)、ensg00000103091(wdr59)和ensg00000140564(furin),其结果是auc值为0.70(95%-ci:0.47-0.94)且分类准确度为73%。
(5)归类癌症存在或者不存在
表2中列出的至少5个基因的基因表达水平可用于从受试者的样品中归类所述受试者中存在或不存在癌症。
为此,从已知不患有癌症(诸如肺癌)的受试者中分离出无核细胞、优选凝血细胞。从所述经分离的无核细胞中分离包含核糖核酸(rna)、优选信使rna(mrna)的样品。在使用本领域技术人员已知的任何方法将rna逆转录成脱氧核糖核酸(cdna)拷贝后,标记所得的cdna并且例如通过下一代测序(例如在illumina测序平台上)定量基因表达水平。
基于测序结果,在来自包含所述癌症患者的核糖核酸(rna)的样品中确定表2中列出的至少5个基因,并且优选标准化。将标准化的表达水平与参考样品中的相同的至少5个基因的表达水平进行比较。所述参考样品从一个或更多个癌症患者获得,和/或从一个或更多个已知不患有癌症的受试者获得。从所述比较,可以归类受试者是否患有癌症(诸如肺癌)的可能性。
在根据本发明的优选方法中,确定表2中的至少5个基因的表达水平,更优选表2中至少10个基因的表达水平,更优选表2中至少20个基因的表达水平,更优选表2中至少30个基因的表达水平,更优选表2中至少40个基因的表达水平,更优选表2中至少50个基因的表达水平,更优选表2中所有上千个基因的rna表达水平。
进一步优选的是,来自表2的所述至少5个基因包含如表2中所示的具有最低p值的前5个基因,更优选如表2所示的具有最低的p值的前10个基因,更优选如表2所示的具有最低p值的前20个基因,更优选如表2中所示的具有最低p值的前30个基因,更优选如表2中所示的具有最低p值的前40个基因,更优选如表2中所示的具有最低p值的前50个基因。
在进一步优选的实施方案中,来自表2的所述至少5个基因包含hbb、eif1、capns1、ndufaf3和otud5,更优选hbb、eif1、capns1、ndufaf3、otud5、srsf2、anp32b、kifap3、atox1和bcap31,更优选hbb、eif1、capns1、ndufaf3、otud5、srsf2、anp32b、kifap3、atox1、bcap31、nap1l1、timp1、polr2e、cd74、polr2g、rps5、gpi、gstm4、ighm和dstn,更优选hbb、eif1、capns1、ndufaf3、otud5、srsf2、anp32b、kifap3、atox1、bcap31、nap1l1、timp1、polr2e、cd74、polr2g、rps5、gpi、gstm4、ighm、dstn、aldh9a1、znf346、lman1、eef1b2、ap2s1、hspb1、hbq1、htatip2、ptms和tpm2,更优选hbb、eif1、capns1、ndufaf3、otud5、srsf2、anp32b、kifap3、atox1、bcap31、nap1l1、timp1、polr2e、cd74、polr2g、rps5、gpi、gstm4、ighm、dstn、aldh9a1、znf346、lman1、eef1b2、ap2s1、hspb1、hbq1、htatip2、ptms、tpm2、desi1、rhoc、ywhah、cpq、mtpn、iscu、mrpl37、mgst3、cmtm5和actg1,更优选hbb、eif1、capns1、ndufaf3、otud5、srsf2、anp32b、kifap3、atox1、bcap31、nap1l1、timp1、polr2e、cd74、polr2g、rps5、gpi、gstm4、ighm、dstn、aldh9a1、znf346、lman1、eef1b2、ap2s1、hspb1、hbq1、htatip2、ptms、tpm2、desi1、rhoc、ywhah、cpq、mtpn、iscu、mrpl37、mgst3、cmtm5、actg1、itga2b、hpse、klhdc8b、cdc37、hla-dra、ksr1、acot7、prkar1b、maob和zdhhc12,更优选hbb、eif1、capns1、ndufaf3、otud5、srsf2、anp32b、kifap3、atox1、bcap31、nap1l1、timp1、polr2e、cd74、polr2g、rps5、gpi、gstm4、ighm、dstn、aldh9a1、znf346、lman1、eef1b2、ap2s1、hspb1、hbq1、htatip2、ptms、tpm2、desi1、rhoc、ywhah、cpq、mtpn、iscu、mrpl37、mgst3、cmtm5、actg1、itga2b、hpse、klhdc8b、cdc37、hla-dra、ksr1、acot7、prkar1b、maob、zdhhc12、snx3、yif1b、prdx5、hdac8、ddx5、tpm1、svip、pdap1、cd79b和prss50,更优选hbb、eif1、capns1、ndufaf3、otud5、srsf2、anp32b、kifap3、atox1、bcap31、nap1l1、timp1、polr2e、cd74、polr2g、rps5、gpi、gstm4、ighm、dstn、aldh9a1、znf346、lman1、eef1b2、ap2s1、hspb1、hbq1、htatip2、ptms、tpm2、desi1、rhoc、ywhah、cpq、mtpn、iscu、mrpl37、mgst3、cmtm5、actg1、itga2b、hpse、klhdc8b、cdc37、hla-dra、ksr1、acot7、prkar1b、maob、zdhhc12、snx3、yif1b、prdx5、hdac8、ddx5、tpm1、svip、pdap1、cd79b、prss50、gpx1、ifitm3、samd14、fundc2、brix1、cfl1、akirin2、napsb、gpaa1、trim28、cmtm3和mmp1。
在最优选的实施方案中,来自表2的所述至少10个基因包含ensg00000168765(gstm4)、ensg00000206549(prss50)、ensg00000106211(hspb1)、ensg00000185909(klhdc8b)、ensg00000097021(acot7)、ensg00000105401(cdc37)、ensg00000099817(polr2e)、ensg00000105220(gpi)、ensg00000075945(kifap3)、ensg00000100418(desi1)。在独立的晚期验证组(n=518个样品)中,10个基因引起auc值为0.74(95%-ci:0.70-0.77),分类准确度为68%。在早期验证组(n=106个样品)中auc值为0.69(95%-ci:0.59-0.79),分类准确度为65%。
在最优选的实施方案中,来自表2的一组至少45个基因用于从受试者的样品归类所述受试者存在或者不存在癌症,尤其是肺癌。所述至少45个基因包含ensg00000023191(rnh1)、ensg00000142089(ifitm3)、ensg00000097021(acot7)、ensg00000172757(cfl1)、ensg00000213465(arl2)、ensg00000136938(anp32b)、ensg00000067365(mettl22)、ensg00000130429(arpc1b)、ensg00000116221(mrpl37)、ensg00000177556(atox1)、ensg00000074695(lman1)、ensg00000188467(tpm2)、ensg00000188191(prkar1b)、ensg00000126247(capns1)、ensg00000159335(ptms)、ensg00000113761(znf346)、ensg00000102265(timp1)、ensg00000168002(polr2g)、ensg00000185825(bcap31)、ensg00000155366(rhoc)、ensg00000099817(polr2e)、ensg00000125868(dstn)、ensg00000160446(zdhhc12)、ensg00000100418(desi1)、ensg00000109854(htatip2)、ensg00000161547(srsf2)、ensg00000068308(otud5)、ensg00000206549(prss50)、ensg00000178057(ndufaf3)、ensg00000042753(ap2s1)、ensg00000168765(gstm4)、ensg00000075945(kifap3)、ensg00000173812(eif1)、ensg00000086506(hbq1)、ensg00000106244(pdap1)、ensg00000187109(nap1l1)、ensg00000106211(hspb1)、ensg00000105220(gpi)、ensg00000105401(cdc37)、ensg00000128245(ywhah)、ensg00000173083(hpse)、ensg00000185909(klhdc8b)、ensg00000126432(prdx5)、ensg00000166091(cmtm5)和ensg00000069535(maob)。在独立的晚期验证组(n=518个样品)中,45个基因引起auc值为0.77(95%-ci:0.73-0.81),分类准确度为77%。在期验证组中的auc值为0.74(95%-ci:0.65-0.83),分类准确度为70%(n=106个样品)。
(6)额外的p-选择素谱。
p选择素(selp,cd62)储存在血小板α-颗粒中并在血小板活化时释放。较年轻的网状血小板富含p-选择素水平。表2中描述的选择用于nsclc诊断的血小板rna基因小组包含与血小板中的p-选择素rna表达共调节的基因。因此,nsclc诊断特征可以富含表达高水平的p-选择素rna的网状血小板。如果响应患者的血小板群在治疗期间从网状血小板转变为成熟血小板,则所述p-选择素特征可有助于预测治疗响应。对于其他治疗模块,包括化学疗法、靶向疗法、放射疗法、手术或免疫疗法,也可以观察到这种转变。
因此,表3中列出的至少5个基因的基因表达水平可用于在施用调节pd-1与其配体之间的相互作用的免疫疗法之前,帮助预测对所述疗法响应。
因此,本发明提供了一种对癌症患者施用调节pd-1与其配体之间的相互作用的免疫疗法的方法,包括提供来自患者的样品的步骤,所述样品包含从所述患者的无核细胞获得的mrna产物;确定表1中列出的至少4个基因,更优选表1中列出的至少5个基因,和表3中列出的至少5个基因的基因表达水平;将所述确定的基因表达水平与参考样品中所述基因的参考表达水平进行比较;根据与参考文献的比较,将患者归为所述免疫治疗的阳性响应者,或归为非阳性响应者;并且对被归为阳性响应者的癌症患者施用免疫疗法。
为此,从已知患有癌症(诸如肺癌)的患者中分离出无核细胞、优选凝血细胞。从分离的无核细胞中分离包含核糖核酸(rna)、优选信使rna(mrna)的样品。在使用本领域技术人员已知的任何方法将rna逆转录成脱氧核糖核酸(cdna)拷贝后,标记所得的cdna并且例如通过下一代测序,例如在illumina测序平台上定量基因表达水平。
基于测序结果,在包含来自所述癌症患者的核糖核酸(rna)的样品中确定表3中列出的至少5个基因的基因表达水平并且优选标准化。将标准化的表达水平与参考样品中相同的至少5个基因的表达水平进行比较。所述参考样品从一个或更多个已知对调节pd-1与其配体之间的相互作用的免疫疗法成阳性响应的癌症患者获得,和/或从一个或更多个已知对调节pd-1与其配体之间的相互作用的免疫疗法成阴性响应的癌症患者获得。从所述比较,获得了预测的对施用调节pd-1与其配体之间的相互作用的免疫疗法(诸如施用nivolumab)的响应效力。
在根据本发明的优选方法中,确定来自表3的至少5个基因的表达水平,更优选来自表3的至少10个基因的表达水平,更优选来自表3的至少20个基因的表达水平,更优选来自表3的中至少30个基因的表达水平,更优选来自表3的至少40个基因的表达水平,更优选来自表3的至少50个基因的表达水平,更优选来自表3的所有1820个基因的rna表达水平。
进一步优选的是,来自表3的所述至少5个基因包含如表3中所示的具有最低p值的前5个基因,更优选如表3所示的具有最低的p值的前10个基因,更优选如表3所示的具有最低p值的前20个基因,更优选如表3中所示的具有最低p值的前30个基因,更优选如表3中所示的具有最低p值的前40个基因,更优选如表3中所示的具有最低p值的前50个基因。
在进一步优选的实施方案中,来自表3的所述至少5个基因包含来自表3的selp、itga2b、ap2s1、otud5和maob,更优选selp、itga2b、ap2s1、otud5、maob、kifap3、hbq1、acot7、polr2e和desi1,更优选selp、itga2b、ap2s1、otud5、maob、kifap3、hbq1、acot7、polr2e、desi1、timp1、cpq、gpi、cdc37、mtpn、hspb1、pdap1、htatip2、snx3和znf346,更优选selp、itga2b、ap2s1、otud5、maob、kifap3、hbq1、acot7、polr2e、desi1、timp1、cpq、gpi、cdc37、mtpn、hspb1、pdap1、htatip2、snx3、znf346、dstn、capns1、prdx5、ywhah、akirin2、iscu、tpm1、cmtm3、aldh9a1和rhoc,更优选selp、itga2b、ap2s1、otud5、maob、kifap3、hbq1、acot7、polr2e、desi1、timp1、cpq、gpi、cdc37、mtpn、hspb1、pdap1、htatip2、snx3、znf346、dstn、capns1、prdx5、ywhah、akirin2、iscu、tpm1、cmtm3、aldh9a1、rhoc、ptms、zdhhc12、srsf2、fundc2、cmtm5、samd14、yif1b、polr2g、gstm4和cfl1。更优选selp、itga2b、ap2s1、otud5、maob、kifap3、hbq1、acot7、polr2e、desi1、timp1、cpq、gpi、cdc37、mtpn、hspb1、pdap1、htatip2、snx3、znf346、dstn、capns1、prdx5、ywhah、akirin2、iscu、tpm1、cmtm3、aldh9a1、rhoc、ptms、zdhhc12、srsf2、fundc2、cmtm5、samd14、yif1b、polr2g、gstm4、cfl1、hpse、eif1、ndufaf3、actg1、bcap31、klhdc8b、nap1l1、prkar1b、mmp1、gpaa1、svip、tpm2、prss50和gpx1。
来自表3的至少5个基因的最优选的组包含ensg00000161203(ap2m1)、ensg00000204420(c6orf25)、ensg00000204592(hla-e)、ensg00000064601(ctsa)和ensg00000005961(itga2b)。使用这组额外的基因(不仅最优选的至少10个基因的组)引起早期nsclc的分类,auc值为0.66(95%-ci:0.55-0.76),准确度为65%(n=106个样品)。
(7)定义粒子群优化
可以利用几种生物信息学优化算法来解决关于参数选择的数学问题。这些优化过程迭代地寻找确定数学问题的参数的最佳参数设置。该迭代过程由优化算法有效且高效地指导。我们声称粒子群智能优化(pso)用于液体活检中基因小组选择的包括参数选择子变量和与其他数学优化算法的杂交/组合的参数选择的数学方法。我们将pso定义为利用使用高维空间中的迭代重定位的粒子位置和粒子速度的元算法,所述pso用于进行有效和优化的参数选择,即基因小组选择。pso还包括可用于自动和增强的基因小组选择的其他优化元算法。我们测试了粒子群优化算法,并证明了增强的pso算法能够从血小板rna-seq文库(n=728)中有效选择剪接rna生物标志物组。这引起iv期非小细胞肺癌(nsclc)的准确的基于tep的检测(n=520独立验证群组,准确度:89%,auc:0.94,95%-ci:0.93-0.96,p<0.001),与个体年龄、全血储存时间和各种炎症情况无关。此外,我们采用群智能来探索剪接rna生物标志物谱,其用于在抗pd-1nivolumab免疫疗法的基线时刻对iv期nsclc患者的基于血液的治疗响应预测(n=64)。nivolumab响应预测算法引起88%的准确度(auc0.89,95%-ci:0.8-1.0,p<0.01)。据我们所知,这是pso首次用于选择生物标志物基因小组以诊断癌症和预测tep治疗响应的示范。利用pso算法优化确定用于支持向量机械训练的基因小组的四个参数。除了分析来自tep的rna分子外,通过对pso算法进行类似或组合的数据输入,pso还可用于分析小rna、rna重排、dna单核苷酸改变、蛋白质水平、代谢组水平,这些成分分离自tep、血浆、血清、循环肿瘤细胞或细胞外囊泡。
出于清楚和简明描述的目的,在此将特征描述为相同或单独实施方案的一部分,然而,应当理解,本发明的范围可以包括具有所描述的全部或一些特征的组合的实施方案。
表1
表2
表3
实施例
实施例1
一般材料和方法
研究设计和样品选择
在荷兰阿姆斯特丹的vu大学医学中心、荷兰阿姆斯特丹的荷兰癌症研究所(nki/avl)、荷兰阿姆斯特丹的学院医学中心、荷兰乌得勒支的乌特勒支医学中心、瑞典于默奥的于默奥大学医院、西班牙巴塞罗那的德国人特利亚斯普约尔医院(thehospitalgermanstriasipujol)、意大利比萨的比萨大学医院和美国波士顿的马萨诸塞州综合医院,通过静脉穿刺从癌症患者、患有炎症和其他非癌病症的患者以及无症状个体抽取外周全血。将全血收集在含有抗凝血剂edta的4、6或10-mledta包被的紫色封盖的bdvacutainer中。癌症患者通过临床、放射学和病理学检查确诊,并且确认在血液采集时可检测到肿瘤负荷。所包括的106个nsclc样品是相同患者的随访样品,在第一次采血后数周至数月收集。使用matlab中的自定义脚本回顾性地进行年龄匹配,通过排除和包括针对两组之间的类似中位年龄和年龄范围的非癌症和nsclc样品来迭代匹配样品。对训练评估和验证群组的样品进行了类似和同时的收集和处理。表4中提供了所包括的样品、人口统计学特征、起源医院、血液采集和血小板分离之间的时间(血液储存时间)以及使用的分析方法和分类器的详细概述。无症状的个体在血液采集,或以前,被诊断为未患有癌症,但没有进行额外的证实没有癌症的测试。患有炎症或其他非癌变病症的患者在采血时没有诊断出恶性肿瘤。该研究是根据赫尔辛基宣言的原则进行的。本研究的批准来自每个参与医院的机构审查委员会和伦理委员会。由于根据医院的道德规则对这些样品进行匿名化,无法获得无症状个体的临床随访。
临床数据注释
对于临床数据的收集和注释,手动查询针对人口统计学变量(即年龄、性别、吸烟、肿瘤类型、转移、当前和先前治疗的细节以及合并症)的患者记录。在跨性别者的情况下,标明了新的性别(n=1)。在(新的)治疗开始之前或治疗期间收集血小板样品、各自的基线和随访样品。在基线、治疗开始后6-8周,3个月和6个月通过ct成像进行用nivolumab治疗的患者的响应评估(图2)。对于nivolumab响应预测算法,在开始治疗之前一个月收集的样品被注释为基线样品。根据已更新的recist1.1版标准评估治疗反应,评分为进行性疾病(pd)、稳定的疾病(sd)、部分响应(pr)或完全响应(cr)(eisenhaueretal.,,2009,europeanjournalofcancer,45:228-247;schwartzetal.,,2016,europeanjournalofcancer62:132-137)。有关详细的示意图,请参见图2a。我们的目的是确定那些治疗对疾病有控制作用的患者。因此,对于nivolumab响应预测分析,我们将患有进行性疾病的患者分组为非响应组中的最佳反应,总共60个样品。在6个月响应评估时,在任何响应评估时间点具有部分响应的患者作为最佳反应或稳定疾病被注释为响应者,总共44个样品。所有临床数据都是匿名的,并存储在安全的数据库中。
混杂变量分析
估计变量1)血液采集时的患者年龄(以年为单位)、2)全血储存时间、3)性别及4)吸烟(当前、以前、从不)的贡献,我们总结了来自我们先前研究的补充表s1a-c和补充图s2c的可用的数据(bestetal.,2015,cancercell,28:666-676),并在统计软件模块sas(v.13.0.0;sasinstituteinc.,100sascampusdrive,cary,nc27513-2414,usa)中进行逻辑回归分析。血液储存时间定义为血液采集和通过差速离心分离血小板开始之间的时间,分为<12小时组和>12小时组。对于缺少数据的样品的变量,从计算中排除特定样品的特定值。通过选择疾病状态作为角色变量y并添加患者年龄、血液储存时间、性别、吸烟和nsclc的预测强度作为模型效应,使用具有名义响应的逻辑回归的测量来评估对nsclc的诊断分类器的患者年龄、血液储存时间和预测强度的联合预测能力。所有其他设置均默认设置。
血液处理和血小板分离
如前所述,在48小时内使用标准化方案处理4ml、6ml或10mledta包被的vacutainer管中的全血样品(bestetal.,2015.cancercell28:666-676;nilssonetal.,2011.blood118:3680-3683)。在vu大学医学中心、荷兰癌症研究所、乌特勒支医疗中心、于默奥大学医院,德国人特利亚斯普约尔医院和比萨大学医院收集的全血在采血后12小时内进行血小板分离。在波士顿马萨诸塞州综合医院和阿姆斯特丹医学中心收集的全血样品存放过夜并在24小时后处理。为了分离血小板,通过20分钟120xg离心步骤将血小板富含的血浆(prp)与有核血细胞分离,之后通过20分钟360xg离心步骤使血小板形成小球。必须小心地去除9/10的prp,以降低有核细胞污染血小板制剂的风险,在淡黄色层中形成小球。在室温下进行离心。将血小板小球小心地重悬于rnalater(lifetechnologies)中,并在4℃下孵育过夜后,在-80℃冷冻。
血小板活化的流式细胞术分析
为了评估我们的血小板分离过程中相对的血小板活化,我们使用bdfacscalibur流式细胞仪测量了组成型表达的血小板标志物cd41(apc抗人,克隆:hip8)和血小板活化依赖性标志物p-选择素(cd62p,pe抗人,克隆:ak4,biolegend)和cd63(fitc抗人,克隆:h5c6,biolegend)的表面蛋白表达水平。我们从6个健康供体中的每一个收集了5个6mledta包被的vacutainer管,并确定了基线(0小时)、8小时、24小时、48小时和72小时的血小板活化状态。作为阴性对照,我们使用从柠檬酸盐抗凝全血的标准化血小板分离方案在零时间点从全血中分离血小板,其已经被验证用于诱导最低的血小板活化。该方案包括收集血小板富含的血浆后,optiprep(sigma)密度梯度离心(350xg,15分钟)的步骤。然后进行两次洗涤步骤,首先用hepes,然后在ssp+(macopharma)缓冲液中进行洗涤步骤。我们在每次离心步骤之前使用400nm前列腺素12(sigma-aldrich)以在分离过程中防止血小板活化。作为阳性对照,我们包括了由20μmtrap(traptest,roche)活化的血小板。血小板小球分离后固定在0.5%甲醛(roth)中,染色,并储存在1%甲醛中用于流式细胞术分析。用flowjo分析相对活化和平均荧光强度(mfi)值。因此,通过p-选择素和cd63血小板表面标志物的稳定水平证实了在血液收集和储存期间不存在血小板活化(图4b)。
总rna分离、smarter扩增和truseq文库制备
用于测序的样品的制备分批进行,并且每批包括临床条件的混合物。对于血小板rna分离,将冷冻的血小板在冰上解冻,并使用mirvanamirna分离试剂盒(ambion,thermoscientific,am1560)分离总rna。将血小板rna在30μl洗脱缓冲液中洗脱。我们使用rna6000picochip(bioanalyzer2100,agilent)评估血小板rna质量,并且作为后续实验的质量标准仅包括rin值>7和/或独特rrna曲线的血小板rna样品。所有bioanalyzer2100质量和数量测量均使用默认设置从自动生成的bioanalyzer结果报告中,并在对参考梯度(数量、外观和坡度)进行严格评估后收集。用于illumina测序的truseqcdna标记(labelling)方案(见下文)需要~1μg的输入cdna。由于单个血小板含有估计的~2飞克的rna(teruel-montoyaetal.,2014.plosone9(7):e102259),假设全血的平均血小板计数为300×106/ml以及高效血小板分离和rna提取,从临床相关血液体积(6ml)估计的血小板最佳产量约为3.6微克。从我们的血液样品中获得的平均总rna是146ng(标准偏差:130ng,n=237个样品,参见图4c)。在非癌症个体(n=86)和nsclc患者(n=151)之间的6mledta包被的vacutainer管中收集的全血的总血小板rna产量的测量引起nsclc患者血小板中总rna的轻微但显著的增加(p=0.0014,学生t检验,图4c),这归因于nsclc患者中血小板更新的潜在差异(也参见实施例3)。为了获得足够的血小板cdna用于稳健的rna-seq文库制备,使用用于illumina测序v3的smarterultralowrna试剂盒(clontech,目录号634853)对样品进行cdna合成和扩增。在扩增之前,将所有样品稀释至~500pg/微升总rna,并再次使用bioanalyzerpicochip确定和定量质量。对于原液产量低于400pg/微升的样品,使用总rna的两倍或更多微升(高达~500pg总rna)的体积作为smarter扩增的输入。使用具有dna高灵敏度芯片(agilent)的bioanalyzer2100测量扩增的cdna的质量控制。所有smartercdna合成和扩增与阴性对照一起进行,其通过bioanalyzer分析需要阴性对照。选择在300-7500bp区域中具有可检测片段的样品用于进一步处理。为了测量平均cdna长度,我们在bioanalyzer软件中选择了200-9000个碱基对的区域并记录了平均长度。为了标记用于测序的血小板cdna,首先通过超声处理(covarisinc)对所有扩增的血小板cdna进行核酸剪切,随后使用truseqnanodna样品制备试剂盒(illumina,catnr.fc-121-4001)用单索引条形码标记用于illumina测序。为了解释低血小板cdna输入浓度,使用15分钟珠子-cdna结合步骤和10循环的富集pcr进行所有珠子清除步骤。所有其他步骤均根据制造商方案。使用dna7500芯片或dna高灵敏度芯片(agilent)测量经标记的血小板dna文库质量和数量。为了使用于smarter扩增的总rna输入、smarter扩增cdna产量和truseqcdna产量(图4d、e)相关,将所有可用的具有匹配数据的样品进行pearson相关性检验(r中的相关检验函数)。以等摩尔浓度合并产物大小在300-500bp之间的高质量样品(每个池12-19个样品),用于浅度thromboseq(shadowthromboseq),并提交用于在使用4型测序试剂illuminahiseq2500平台上进行的100bpsingleread测序。对于深度thromboseq(deepthromboseq)实验(参见图41),我们合并了12个相同制备的血小板样品,并在hiseq2500流动池的四个泳道上对相同的池进行测序。随后,每个样品的四个单独的fastq文件在计算机中合并。
原始rna测序数据的处理
如先前所述(bestetal.,2015.cancercell28:666-676),将在fastq文件中编码的血小板的原始rna序列数据进行标准化的rna-seq比对管线。总之,通过trimmomatic(v.0.22)(bolgeretal.,2014.bioinformatics30:2114-2120)对rna序列读数进行序列衔接子的修整和裁剪,使用star(v.2.3.0)定位到人参考基因组(hg19)(dobinetal.,2013.bioinformatics29:15-21),并使用由ensembl基因注释版本75指导的htseq(v.0.6.1)进行总结(andersetal.,2014.bioinformatics31:166-169)。所有后续的统计和分析性分析均在r(版本3.3.0)和r-studio(版本0.99.902)中进行。在测序后总共产生少于0.2x10e6跨越内含子的读数的样品中,我们再次测序样品的原始truseq制剂并在htseq计数总结后合并从两个单独的fastq文件产生的读数计数(对n=52样品进行)。除了图6b中的分析之外,在线粒体dna和y染色体上编码的基因被排除在下游分析之外。正如预期的那样,在对经多腺苷酸化rna进行测序后,我们测量到了映射到外显子区域的血小板序列读数的显著富集(图6b)。通过评估文库复杂性进行样品筛选,文库复杂性与跨越内含子的读数文库大小部分相关(图4j)。首先,对于所有测序的血小板样品,我们排除了在>90%的群组中产生<30个跨越内含子的读数的基因(总共n=740,n=385非癌症和n=355nsclc)。这产生了检测到具有足够的覆盖度的4722个不同基因的血小板rna-seq文库。对于每个样品,我们定量了用于定位至少一个跨越内含子的读数的基因数量,并且排除了具有<3000个检测到的基因的样品(约1%下限,图4j)。因此,我们排除了10个样品(n=8(总数的2.1%)非癌症,n=2(总数的0.6%)nsclc)。接下来,为了排除显示低样品间相关性的血小板样品,我们进行了留一样品法的互相关分析(图4m)。在数据标准化之后(参见实施例1中的“数据标准化和ruv介导的因子校正”部分),对于群组中的每个样品,除了“测试样品”之外的所有样品用于计算每个基因的中位每百万计数表达(参考谱)。接下来,测试样品与参考集的可比性由pearson相关性确定。排除相关性<0.5的样品(n=2),且剩余的728个样品包括在该研究中(图1a)。值得注意的是,我们观察到bioanalyzercdna谱(突起的/平滑的模式)的微妙差异,与患者组无关,但与平均cdna长度显著相关(图4f、g)。在实施例2中更详细地讨论了该观察结果。我们使用bedtools(v2.17.0,在bedtools交叉后bedtools合并)分别测量了针对突起和平滑样品的映射到基因间区域的连锁读数的平均长度,并且观察到大多数读数(突起样品>10.9%,平滑样品>13.5%,每个n=50个样品)的平均片段长度(连接读数)<250nt,峰值在100-200nt。我们将cdna谱的差异部分归因于血小板分离过程中保留的“污染”血浆dna(图4h和实施例2)。为了防止潜在的血浆dna参与我们的计算血小板rna分析,我们只选择了剪接的跨越内含子的rna读数(图1b,图4i)。
thromboseq的技术性能的评估
我们在血小板rna中观察到丰富的剪接rna库(图4k),包括4000-5000种不同的信使rna和非编码rna。剪接的血小板rna多样性与先前对血小板rna谱的观察结果一致(bestetal.,2015.cancercell28:666-676;rowleyetal.,2011.blood118:e101-11;brayetal.,2013.bmcgenomics14:1;gnatenkoetal.,2003.blood101:2285-2293)。为了评估从~500pg总血小板rna输入检测4000-5000个血小板rna库的效率(图4k),我们总结了具有至少30个非标准化跨越内含子的读数计数的所有基因标签。我们研究了收集更多单读100bprna-seq读数(约5倍深度:深度thromboseq)的血小板cdna文库(n=12个健康供体)是否在检测到更低丰度的rna时产生(图41)。为此,我们选择了至少一个样品中具有超过10个原始跨越内含子的读数的基因标签。这是针对浅度和深度thromboseq分别进行的。出于可视化目的,我们计算了中位原始跨越内含子的读数计数,对计数进行了对数转换(在向所有标签添加一个计数之后),并绘制了具有最高计数数量的20000个基因标签。同样,这是针对浅度和深度thromboseq数据分别进行的。增加浅度thromboseq的平均覆盖率seq~5x不会产生显著富集的低丰度血小板基因检测。
差异化剪接分析
在差异化剪接分析之前,数据经历如实施例1中的“数据标准化和ruv介导的因子校正”部分所述的迭代校正模块(年龄相关阈值0.2,文库大小相关阈值0.8(非癌症/nsclc),图5a)或0.95(nivolumab治疗响应特征,图4b))。经校正的读数计数被转换为每百万计数,对数转换,并乘以由r-packageedger的calcnormfactors函数计算的tmm标准化因子(robinsonetal.,2010.bioinformatics26:139-140)。为了产生差异化剪接基因组,获得负二项式模型的后拟合以及常见的、有标签的和趋势分散估计,使用广义线性模型(glm)似然比检验确定差异化表达的转录本,如在edger-package中实施的。出于数据信号的目的,我们使用校正的读数计数作为输入,用事后基因本体解释进行用于差异化剪接分析的差异化表达分析,而对于分类任务期间数据的再现性,我们使用未校正的原始读数计数作为输入。从剪接rna基因列表中除去每百万具有少于三个对数计数的基因(logcpm)。具有用于多重假设检验(fdr)低于0.01的校正的p值的rna被认为是统计学显著的。对于使用差异化剪接分析(图2b)和分类算法(图2c)的nivolumab响应预测特征开发,我们使用p值统计进行基因选择。使用列系统树图(ward聚类)的fisher精确检验计算的p值作为性能参数(另见在实施例1中“群增强的thromboseq算法的性能测量”部分),nivolumab响应预测特征阈值由群智能确定。通过ward聚类和pearson距离进行热图行和列系统树图的无监督层次聚类。使用fisher精确检验(r中fisher.test-函数)确定无监督层次聚类的非随机分区和相应p值。为了确定非癌症个体和nsclc患者血小板之间的差异化剪接水平(图5),我们仅包括用患者年龄及血液储存时间匹配群组而分配的样品(总共训练和验证,n=263,还参见图3c和4a)。
rna-seq读取分布的分析
在用患者年龄及血液储存时间匹配的nsclc/非癌症群组(训练、评估和验证,总共263个样品)而分配的样品中研究了血小板cdna的映射rna-seq读数的分布,并从而研究了rna片段的来源。分别定量线粒体基因组和人类基因组(后者包括外显子、内含子和基因间区域)(图6a)。使用samtoolsview算法(v.1.2,选项-q30,-启用c)进行读取定量。对于外显子读数的定量,我们仅通过在samtoolsview定量之前进行bedtoolsintersect筛选步骤(-abam,-wa,-f1,v.2.17.0)来选择完全映射到外显子的读数。我们使用ensembl基因注释版本37中注释的外显子、内含子和基因间区域和hg19的床文件作为参考。通过选择bamn文件中的雪茄标签(cigar-tag)从比对的读数中筛选剪接rna,并通过仅定量映射到′chrm′的读数来选择映射到线粒体基因组的读数。我们确定了通过计算读数比例的映射到特定基因组区域的读数与每个样品的定量读数总数相比的比率。使用r中的t检验函数进行独立学生t检验。实施例3中提供了结果和数据解释的详细描述。
p-选择素特征(p-selectinsignature)
为了确定p选择素水平和外显子读数计数之间的相关性,我们比较了263名患者年龄及血液储存时间匹配的个体的p-选择素(selp,ensg00000174175)每百万计数值与映射到外显子的读数的数量(图7a)。从log2转化的,tmm标准化的和每百万计数转化的读数计数,进行ruv介导的校正收集p-选择素表达水平(参见实施例1中的“数据标准化和ruv介导的因子校正”部分,年龄相关阈值0.2,库大小相关阈值0.9)。对p-选择素表达水平的外显子读数计数使用pearson相关性进行相关性分析。为了鉴定与p-选择素富集相关的基因表达,我们计算了所有个体基因(总共n=4722)对p-选择素表达水平的pearson相关性。数据在直方图中总结,并且我们通过选择正的(r>0)和最显著(fdr<0.01,针对多重假设检验调整)相关基因来编辑p-选择素特征。将p-选择素特征与非癌症和nsclc之间的所有差异和越来越多的剪接基因进行比较(图5a),并在维恩图(r中的venndiagram-package)中进行总结。
可变剪接异构体和外显子跳跃事件分析
在我们的100bp单读rna-seq数据中对可变剪接分析,我们采用了miso算法(katzetal.,2010.naturemethods7:1009-15)。简而言之,miso算法定量了有利于包含或排除特定注释事件(诸如外显子跳跃或rna异构体)的读数的数量。通过对支持一种变体或另一种(开/关)的读数和支持两种异构体的读数进行评分,该算法推断包含率,从而推断出剪接的百分比(psi)。在实施例3中提供了tep中可变剪接分析的详细描述和结果的解释。
用于miso剪接分析的原始mrna测序数据的处理
对于misorna剪接分析(图6c和d)中,患者年龄及血液储存时间匹配的nsclc/非癌症群组的fastq-文件再次进行trimmomatic修整和裁剪,以及star读取映射(也参见实施例1中的“原始rna测序数据的处理”部分)。为了创建所有已输入读数的统一读取长度,如miso算法所要求的,将修整的读数裁剪为92bp,并且从分析中排除读取长度为92bp以下的读数。另外使用picard工具(添加或替代读数组函数,v.1.115,)读取组后,进行misosam-到-bam转换,并且所述索引bam文件进行使用hg19和索引ensembl基因注释版本65作为参考的miso算法(v.0.5.3)。使用summarize_miso-函数总结miso输出文件。随后使用matlab中的自定义脚本将经总结的异构体和跳跃的外显子的miso文件转换为“psi”计数矩阵和“分配计数”计数矩阵。
鉴定可变剪接的异构体
对于可变异构体分析,我们将分析缩小到血小板中鉴定有确信的跨越内含子的表达水平的4722个基因(参见实施例1中的“原始rna测序数据的处理”部分)。对于miso总结输出文件中可用的每个带注释的ensemble转录本id,已分配的读数计数(分配给特定rna异构体的读数)总结在计数矩阵中。该过程的示意图概述如图6c所示。为了确保正确检测异构体,我们排除了在>90%的样品群组中有<10个读数的rna异构体,并应用了tmm-和每百万计数标准化。接下来,进行已注释的ensembl转录物之间的差异化表达分析,并选择最显著的命中(fdr<0.01,logcpm>1)。有关差异化表达分析的详细信息,请参见实施例1中的“差异化剪接分析”部分。为了鉴定每个亲本基因座的多个rna异构体,我们将ensembl转录物id(enst)与ensembl基因id(ensg)进行匹配并对显著的enst-标签计算ensg-标签的频率度量。通过包括每个亲本基因座的所有enst标签,并比较非癌症和nsclc样品的中位表达值来评估可变剪接的异构体的分布。在所有情况下显示的增加或减少水平的异构体被评分为非可变剪接。在任一组中表现出富集但在另一组中表现出减少,并且对于至少一种其他异构体而言相反的异构体,被评分为可变剪接rna。
外显子跳跃事件的鉴定
为了分析外显子跳跃事件,我们开发了一个定制分析管道,总结了支持包含或排除已注释外显子的读数,并对感兴趣的组(即非癌症与nsclc)的相对贡献进行评分。该算法的输入是psi值计数矩阵和“分配计数”计数矩阵,由miso生成的汇总输出文件生成。前一个计数矩阵需要计算每组的相对psi值和分布,后一个计数矩阵只需要包含rna-seq数据中具有足够覆盖率的外显子(即>60%的样品中>10个读数,其同时支持变体的包含(1,0)和排除(0,1),另见katzetal.,)。覆盖选择器将可用外显子缩减为230个外显子的分析(图6d)。为了选择跳跃外显子事件的差异水平,使用独立学生的t检验比较非癌症和nsclc的psi值,包括事后错误发现率(fdr)校正(在r中t.检验和p.adjust函数)。fdr<0.01的事件被认为是潜在的跳跃外显子事件。通过从中位psi值nsclc中减去每个跳跃事件中的非癌症的中位psi值来计算deltapsi值。
rna结合蛋白基序富集分析-rbp-thrombo搜索引擎
为了鉴定与nsclc患者中的tep特征相关的rna结合蛋白(rbp)谱(图8),我们设计并开发了rbp-thrombo搜索引擎。该算法的基本原理是基因的非翻译区(utr)中特定rbp的富集结合位点与该特定rna的剪接的稳定或调节相关。该算法鉴定了在血小板中可靠地鉴定的基因的基因组utr序列中匹配的rbp结合基序的数目。随后,它将每个包含的rbpn个结合位点与每个单独基因的对数倍数变化(logfc)相关联,并且显著相关性被列为可能涉及的rbp。对于该分析,我们从文献中收集了先前充分表征的rbp结合基序(rayetal.,2013.nature499:.172-177)。该算法利用以下假设:1)utr区域中特定rbp的更多结合位点通过前mrna分子的稳定化或去稳定化预测基因的调节增加(oikonomouetal.,2014.cellreports7:281-292)、2)1)中的功能主要由单个rbp驱动,而不是与多个rbp或mirna或其他顺式或反式调节元件组合或协同作用,和3)包含的rbp存在于非癌症个体和/或nsclc患者的血小板中。为了确定n个rbp结合位点-logfc相关性,该算法进行以下计算和质量测量步骤:
(i)该算法选择所有输入基因的已注释的rna异构体,并鉴定与5′-utr或3′-utr相关的已注释rna异构体的基因组区域。使用bedtools中的getfasta函数(v.2.17.0)从人hg19参考基因组中提取基因组编码序列。在本研究中,我们使用了ensembl注释版本75。
(ii)根据iupac基序注释,从文献中提取的所有经表征的基序序列(总共102个,ray等人的补充表3,(rayetal.,2013.nature499:172-177),对人类进行筛选)减少至547个非冗余(′a′、′g′、′c′和′t′序列)注释。这些非冗余基序序列用作初始搜索的代表性基序序列。
(iii)以迭代方式,每rbp相关的非冗余rbp基序序列与所有已鉴定和已包含的utr序列匹配(使用r中seqinr包的str_计数函数)。
(iv)该算法使用samtoolsview鉴定每个样品映射到每个utr区域的读取数(q30,-启用c,图8b)。没有覆盖或覆盖最小的utr序列被认为在血小板中是不存在的。为了解释由oligo-dt引发的mrna扩增引入的最小偏差(
(v)对于具有与相同亲本基因(ensg)相关的足够覆盖度的所有5′-和3′-utr,将所有匹配的utr-非冗余基序命中相加,并总结在基因-基序矩阵中。通过覆盖所有可能的rbp-基序匹配,将非冗余基序转换为rbp-ids。该矩阵用于下游分析、数据解释和可视化。
我们确认了特定rbp的3′-和5′-utr富集(图8d),并观察到共同参与的rbp的utr-簇(图8e、f)。使用pearson相关性确定对所有rbp的logfc和n个rbp结合位点之间的相关性,并在火山图中总结(图8g)。有关结果的详细描述和解释,请参见实施例4。
数据标准化和ruv介导的因子校正
我们鉴定了两个可能影响分类器预测强度的变量,即血液储存时间和患者年龄(表4)。为了减少参与分类模型的混杂因子的影响,我们应用以下新方法进行迭代rna测序数据校正(也参见图9a中的示意图)。校正模块基于risso等人提出的去除不需要的变量(ruv)方法(rissoetal.,2014.naturebiotech32:896-902;peixotoetal.,2015.nucleicacidsres43:7664-7674),补充了“稳定基因”的选择(独立于混杂变量),以及一种迭代和自动的方法,用于分别去除和包含不需要的和想要的变量。ruv校正方法利用广义线性模型,并使用奇值分解来评估感兴趣的协变量和不想要的变量的贡献(rissoetal.,2014,naturebiotech32:896-902)。原则上,这种方法适用于任何rna-seq数据集,并允许并行地研究许多潜在的混杂变量。值得注意的是,迭代校正算法对于特定样品所属的组(在这种情况下是nsclc或非癌症)是不可知的,并且必要的稳定基因小组仅通过训练群组中包括的样品计算。该算法执行以下多个筛选、选择和标准化步骤,即:
(i)筛选低丰度的基因,即在90%以上的样品群组中少于30个跨越内含子的的剪接rna读数(也包括在一般qc模块中,参见“原始rna测序数据的处理”部分)。
(ii)确定在混杂变量中表现出最小可变性的基因。为此,通过(i)中初始筛选器的每个基因的非标准化原始读数计数使用pearson相关性与总跨越内含子的文库大小(由r中edger包的dgelist函数计算)或个体的年龄相关。具有高pearson相关性(朝向1)的基因显示在每百万计数标准化后的最小可变性(参见图9b、c),并因此被指定为稳定基因。
(iii)训练群组的原始读数计数经历来自r中的ruvseq-包的ruvg-函数。在混杂变量中鉴定的稳定基因被用作“阴性对照基因”。接下来,由ruvg鉴定的每个样品的个体估计因子与潜在的混杂因子(在当前研究中:文库大小、个体年龄)或感兴趣的组(例如非癌症与nsclc)相关。连续(混杂)变量与样品的估计方差相关。使用学生t检验比较二分变量(例如组)。在这两种情况下,p值被用作ruvg变量和(混杂)变量之间的显著替代。值得注意的是,为了防止除去可能与组相关的变量,我们在将变量与(混杂)因子匹配之前应用了两个规则,即a)ruvg变量和组之间的p值应至少>1e-5,和b)ruvg变量和另一个变量之间的p值应至少<0.01。如果该变量与混杂因子相关,则对ruvg变量x校正原始非标准化读数。最后,通过计算每个样品的ruvg校正的读数计数的总和来调整每个样品的总跨越内含子的文库大小。
(iv)使用tmm标准化因子对ruvg标准化的读数计数进行每百万计数标准化、对数变换和乘法。后一标准化因子是使用自定义函数计算的,该函数是从r中edger包中的calcnormfactors函数实现的。这里,tmm参考样品选择的合格样品可以缩小到该群组的子集,即本研究中分配给训练群组的样品,并锁定所选的参考样品。
我们将此迭代校正模块应用于此项工作中的所有分析。估计的不需要变量(k)的因子的ruvg数量是3。我们使用相对对数强度(rle)图直接比较了我们先前的标准化模块和本研究中呈现的迭代校正模块的性能(图9d),并观察到在表达数据中更好地去除变量。使用edaseq包的plotrle函数生成rle图。对于具有和不具有ruv介导的因子校正的每个样品,通过计算样品的中位rle计数与所有样品的总体中位rle计数的绝对差异来确定样品间变异性降低的显著性(图9d)。
基于支持向量机(svm)的算法开发和粒子群驱动的svm参数优化
群增强的thromboseq算法相对于先前公布的thromboseq算法实现了多种改进(bestetal.,2015.cancercell28:666-676)。图9e中提供了群增强的thromboseq分类算法的概述。首先,我们通过实施训练评估方法来改进算法优化和训练评估。将用于匹配群组的总共93个样品(图1d)和用于训练评估的完整群组(图1e)的120个样品用作内部训练群组。这些样品用作迭代校正模块(参见实施例1中的′数据归一化和ruv介导的因子校正′部分)、通过似然比anova检验选择初始基因小组(参见实施例1中的′差异化剪接分析′)、svm参数优化,以及最终算法训练和锁定(支持向量的选择)的参考样品。其次,在似然比anova分析之后,我们去除了具有高内部相关性的基因(在r-包插入符中找到相关函数),因为这些先前被认为在svm模型中导致不想要的噪声。第三,我们实现了先前由guyon等人提出的递归特征消除(rfe)算法(guyonetal.,2002.machinelearning46:389-422),以富集基因小组中最相关且最有助于svm分类器的基因。第四,在最终的svm成本和伽马参数网格搜索之后(参见图9e),我们通过启用内部的第二粒子群算法(在r-packageoptunity中的cv.particle_群体-函数)对成本和伽玛参数进行了额外的细化。为了获得更优的内部svm性能,该内部粒子群算法用于研究和查明由svm网格搜索确定的最佳伽马和成本参数的相邻值。第五,整个svm分类算法经历了由r中的ppso包(optim_ppso_robust-function)实现的粒子群优化算法(pso)(tolsonandshoemaker,2007.waterresourcesresearch43:w01413)。粒子群智能是基于寻求问题的最佳解决方案的搜索空间中粒子的位置和速度。在基于其局部最佳解决方案和总体最佳解决方案对粒子进行迭代重新校准时,可以实现对输入参数和算法设置的更精确估计(图1c)。所实现的算法使得粒子群能够实时可视化,并行地优化多个参数,以及使用多个计算核来部署迭代“函数调用”,从而在大型计算机簇上推进大分类器的实现。pso算法旨在最小化′1-auc′得分。我们采用匹配的nsclc/非癌症群组分类器100粒子进行10次迭代,并使用完整的nsclc/非癌症群组分类器200粒子进行7次迭代。我们优化了通用分类算法的四个步骤,即(i)用于选择在文库大小中鉴定为稳定基因的基因的迭代校正模块阈值(参见图9a),(ii)包括在应用于似然anova检验的结果的差异化剪接筛选器中的fdr阈值,(iii)排除在似然anova检验后选择的高度相关基因,和(iv)通过rfe算法的基因数量。对于本研究中呈现的每个分类任务,将预定义的范围提交给pso算法。svm算法的训练使用两次内部交叉验证,以及用于网格搜索的初始伽马和成本参数范围分别为2^(-20∶0)和2^(0∶20)。为了解释验证群组中未检测到的基因,可能妨碍数据的正常化并降低算法性能,计数在0到12(匹配群组)和0到2(完整群组)之间的基因被训练群组的中位计数替换为特别的基因。
群增强的thromboseq算法的性能测量
我们使用多个训练、评估和验证群组评估了群增强的thromboseq平台的性能、稳定性和可重复性。图3b中提供了用于评估在患者年龄及血液储存匹配群组中的平台性能的群组的示意图。表5中提供了用于分类和分配到不同群组的样品的详细描述。群组的人口统计学和临床特征总结在表4、图4a和表5中。所有分类实验均使用粒子群智能优化的参数用群增强的thromboseq算法进行。我们为匹配的群组(图1d)分配了133个用于训练评估的样品,其中93个用于ruv校正、基因小组选择和svm训练,以及40个用于基因小组优化。完整群组(图1e)包含208个用于训练评估的样品,其中120个用于ruv校正、基因小组选择和smv训练,以及88个用于基因小组优化。nivolumab响应预测群组包含随机样品群组,其由60个训练样品、21个评估样品和23个独立验证样品组成。所有随机选择程序均使用r中实施的样品函数进行。为了将每个群组的样品分配到训练和评估子群组,仅每个临床组的样品数平衡,而其他潜在贡献变量未在此阶段分层(假设各组之间随机分布)。通过留一法交叉验证方法(loocv,也参见best等人(bestetal.,2015.cancercell28:666-676))评估训练群组的性能。在loocv方法期间,所有样品减去一个(“剩余样品”)用于训练算法,之后对剩余样品的响应状态进行分类。每个样品预测一次,引起与训练群组中的样品数量相同的预测。初始训练群组中的稳定基因列表、确定的ruv-去除因子和通过训练评估群组的群体优化确定的最终基因小组用作loocv程序的输入。作为内部再现性的对照,我们随机抽样训练和评估群组,同时保持原始分类器的验证群组和群体引导基因小组,并进行100(nivolumab响应预测)或1000(匹配的和完整群组nsclc/非癌症)训练和分类程序。作为随机分类的对照,用于训练支持向量的svm算法的样品的分类标签被随机排列,同时保持原始分类器的群体引导基因列表。对于已匹配的和完整的nsclc/非癌症群组分类器,该过程进行1000,对于nivolumab响应预测分类器,该过程进行100。如前所述(bestetal.,2015.cancercell28:666-676),相应地计算p值。结果以接收器操作特征(roc)曲线表示,并使用曲线下面积(auc)-值进行概括,如r中的rocr包所确定的。auc95%置信区间根据delong使用的方法用r中proc-包的ci.auc-函数计算(delongetal.,1988.biometrics44:837-45)。
基因本体分析
对于基因本体分析,我们使用scder-package1.99版本(http://pklab.med.harvard.edu/scde/)中执行的pagoda函数研究了共同关联的基因簇。pagoda使得能够通过途径和基因组过度分散分析来冗余异质性模式的聚类和新生基因簇的鉴定(fanetal.,2016.naturemethods13:241-244)。特别地,鉴定新生基因簇的能力对于血小板rna-seq数据的分析是有意义的,因为血小板生物学功能可能未被注释并且只能通过无偏的聚类分析来推断。通过差异化剪接分析(n=1622,图5a)选择的基因id用作生成基因本体库文件的输入。我们使用0.9的距离阈值来减少pagoda冗余,并且启用了新生基因选项的鉴定。分析中的剩余步骤根据pagoda作者的指示。pagoda分析揭示了与疾病状态相关的共调节基因的四个主要簇(一个存在的和三个新生基因簇)。我们选择了具有显著丰富的多重假设检验校正z分数(调整后的z分数)的聚类。2016年9月26日,使用panther分类系统(http://pantherdb.org/)手动进一步策划了新生簇。
实施例2
通过分析smarter扩增后的血小板rna样品,我们观察到smartercdna谱的微妙差异(图4f),正如通过bioanalyzerdna高灵敏度芯片所测量的。cdna产物的斜率可以细分为突起的、平滑的和中间级突起的/平滑的轮廓,并且不倾向于疾病特异性(图4g)。最丰富地观察到斜率的突起的模式(在非癌症中作为nsclc群组的59%)可能与血小板中rna分子的相对小的多样性(测量的约4000-5000个不同的rna)有关。剩余的样品的特征在于平滑的或中间级突起的/平滑的cdna产物谱。值得注意的是,picochiprna谱和dna7500truseqcdna谱在三个smarter组中相似(图4f),并且没有一个smarter组富含低质量rna样品。平均cdna长度可与smarter谱相关,而smarter扩增后的cdna产量相当。值得注意的是,具有更平滑样式的样品引起跨越内含子的剪接rna读数的总计数减少,并且伴随着映射至基因间区域的读数的增加(图4i)。映射到基因间区域的rna-seq读取被认为是源自未注释的基因,导致多个(剪接)读数的堆叠,或(基因组)dna污染导致分散的读取。通过分析基因间区域的小区域(每个1kb),我们观察到这些读数中的少数可归因于潜在的未注释基因(数据未显示)。分析映射到基因间区域的连锁读取片段的平均长度分布(参见实施例1),显示中位片段大小为~100-200bp,在100bp处具有明显的峰,其可能源自无细胞dna的片段(图4h)(newmanetal.,2014.naturemed20:548-554;jiangandlo,2016,trendsgen32:360-371)。我们以前估计了有核细胞在血小板分离过程中的贡献(n=7个随机选择的血小板分离),潜在地解释了基因组dna的痕迹,但仅观察到这些有核(白血)细胞的轻微污染(bestetal.,2015.cancercell28:666-676)。值得注意的是,全血采集和血小板分离程序开始之间的时间可能与smartercdna斜率相关。已经作为全血储存超过24小时的样品在几乎所有情况下都显示出突起的模式,而在采血后直接分离的血小板在大多数情况下显示出平滑的模式。在edta包被的管中收集的全血中无细胞dna相当不稳定,并且在孵育超过12-24小时后,大多数无细胞dna的痕迹可能会降解。因此,我们预计经历血小板分离方案的全血样品-在血液采集后立即或在12小时内-可能被残留的血浆衍生的无细胞dna污染,其痕迹保留在经分离的血小板小球中。通过选择跨越内含子的rna-seq读数,可以避免血小板rna谱中“不需要的”无细胞dna的污染,因为外显子-外显子读数是特异性rna衍生的。因此建议通过在采血后4-24小时内开始血小板分离来标准化样品采集。
实施例3
rna-seq数据提供了以高分辨率定量转录组的几乎任何区域的机会。因此,我们研究了血小板rna谱中rna种类的分布。本研究中分析的血小板构成血液采集时血液中循环的所有血小板的快照,可能受诸如血小板总计数、药物、出血性疾病、损伤、活动或运动以及昼夜节律等变量的影响。对于以下分析,为了减少高度怀疑混淆血小板特征的因子的影响(表4),我们选择了263名患者年龄及血液储存时间匹配的个体。基于内含子读数计数分析,我们鉴定了1625个剪接的血小板基因,具有显著差异化剪接水平(fdr<0.01,nsclc患者的血小板中698个基因具有增强的剪接,以及nsclc患者的血小板中927个基因具有减少的剪接),与先前的发现一致(bestetal.,2015.cancercell28:666-676;calverleyetal.,2010.clinicalandtranslscience3:227-232)。
基于跨越内含子的读数的无监督层次聚类,将非癌症和nsclc样品分成两个不同的组(p<0.0001,fisher精确检验,图5a)。接下来,我们定量了线粒体基因组和人类基因组的每个独立区域(即外显子、内含子和基因间部分)的可信映射的rna-seq读数的数量(参见实施例1)。我们观察到与无癌症个体相比,在nsclc患者中映射到线粒体基因组的读数的平均增加(图6b)。随访分析显示,在nsclc患者中,映射到外显子部分的标准化读数(每百万总基因组读数的读数)的数量增加,而对于内含子和基因间部分,观察到相反的情况(图6b)。我们进一步观察到,对于具有较大比例的映射为跨越内含子的剪接rna读数的读数的样品,映射到线粒体基因组和基因间区域的读数的贡献较低,而具有低跨越内含子的剪接rna读数的样品显示相反的(图4i和6b)。
接下来,我们研究了可变剪接事件对血小板rna库的贡献,因为可变剪接事件可能影响用于诊断分类器的剪接rna读数的数量。为了表征转录组范围内的可变异构体和剪接事件,我们实施了先前公布的用于定量和概括注释的rna异构体的miso算法(katzetal.,2010.naturemethods7:1009-1015)。由此,我们推断出计数矩阵,其包含支持各自包含rna异构体的每个样品的读数(图6c,参见实施例1以获得更多细节)。接下来,我们在rna异构体之间进行差异化表达分析,并在非癌症个体(n=104)和nsclc患者(n=159)之间选择差异化rna异构体。非癌症个体和nsclc患者之间的差异化rna异构体分析显示,在nsclc患者的tep中,743种rna异构体显著富集(n:359)或耗尽(n=384)。在20%(113/571)的基因中,我们鉴定了与相同基因座相关的多种异构体(图6c)。然而,仅在13/571(2.3%)的基因中,我们观察到异构体的潜在可变剪接,尽管这些特定rna异构体之间的差异很小(数据未显示)。总之,这些结果表明,可变剪接的rna异构体仅对tep谱具有轻微至中度的贡献(图1b)。
接下来,我们研究了基因内的可变剪接事件,即外显子跳跃。在这里,我们再次应用miso算法(katzetal.,2010.naturemethods7:1009-1015)来分析38327个已注释的外显子,并推断与相邻外显子相比支持包含或排除特定外显子的读数比例(图6d中的示意图)。此外,该算法为每个事件提供百分比拼接的(psi)值,定量支持包含或排除特定外显子的读数的估计部分。对于外显子跳跃分析,在筛选低覆盖率的外显子后,230个外显子仍然有资格进行分析。我们对每个包含的外显子应用了anova统计,包括多重假设检验(fdr)的校正。通过应用阈值(anovafdr<0.01),我们确定了27个外显子跳跃事件,这些事件在非癌症和nsclc样品之间在psi值上有统计学显著差异(在非癌症中跳跃n=15,在nsclc中跳跃n=12),并且我们观察到nsclc中包含外显子的一般趋势(图6d)。推定的外显子跳跃事件存在于类似snhg6、cd74和srp9的基因中(图6d)。因此,对血小板中可变剪接的分析表明对tep剪接谱的轻微至中度贡献(图1b)。
我们还观察到多个变量收敛,即1)nsclc患者的血小板平均具有较高的rna产量(图4c)、2)nsclc患者的血小板平均显示较低的加工和剪接rna多样性,表明活性降低(图4k),和5)nsclc患者血小板显示增加的映射到外显子和跨越内含子读数的读数表达(图6b),而跨越外显子边界的读数(潜在的未剪接的rna)在非癌症和nsclc中具有相似的水平。符合这些发现,并得到文献报道的支持(dymicka-piekarskaandkemona,2008.thrombosisres122:141-143;dymicka-piekarskaetal.,2006.advancesmedsciences51:304-308;stoneetal.,2012.newenglandjmed366:610-618;watrowskietal.,2016.tumourbiol37:12079-12087),癌症患者的血小板部分似乎富含较年轻的网状血小板。网状血小板是新生血小板(<1日龄),并且含有相当丰富的rna水平,如通过噻唑橙染色所测量的(hoffmann,2014.clinicalchemlabmed52:1107-1117;harrisonetal.,1997.platelets,8:379-383;ingramandcoopersmith,1969.britishjhaematol17:225-229)。估计网状血小板具有20-40倍的富集rna含量(angénieuxetal.,2016.plosone11:e0148064)。因此,我们假设nsclc患者的血小板rna可以富含与较年轻血小板相关的rna,包括p-选择素(cd62)(bernlochneretal.,2016.platelets27:796-804)。我们确实观察到外显子读数覆盖率与p-选择素rna-seq读数计数之间的高度显著的正相关性(n=263,r=0.51,p<0.0001,pearson相关性,图7a)。接下来,我们计算了与p-选择素相关的rna特征,并定义了一个确信检测到并和p-选择素共相关的2797个基因的谱(fdr<0.01,图7b)。p-选择素特征富集了类似casp3(先前涉及巨核细胞介导的促血小板形成(morishimaandnakanishi,2016.genescells21:798-806))、mmp1和timp1(之前显示用于分类血小板(cecchettietal.,2011.blood118:1903-1911))和actb(先前在网状血小板中检测到(angénieuxetal.,2016.plosone11:e0148064))等标志物,提供了p-选择素网状血小板特征的有效性。我们观察到p-选择素特征中77%的基因也被鉴定为在nsclc患者的tep中显著富集(图7c)。因此,我们估计较年轻的网状血小板对nsclc患者的teprna谱的贡献是显著的(图1b和图7c)。
实施例4
血小板是无核细胞碎片。然而,它们含有功能性剪接体和几种剪接因子蛋白(denisetal.,2005.cell122:379-391)。因此,血小板保留其启动前mrna剪接的能力。几个实验已经证明血小板能够根据环境队列剪接前mrna(rondinaetal.,2011.journalthrombhaemostasis9:748-758;schwertzetal.,2006.jexpmed203:2433-2340;denisetal.,2005.cell122:379-391),并且它们具有将rna翻译成蛋白质的能力(weyrichetal.,1998.proceedingsofthenationalacademyofsciences95:5556-5561)。由于血小板缺乏细胞核,但用~20-40飞克的rna包装(angénieuxetal.,2016.plosone11:e0148064)并循环7-10天,(前)mrna需要适当策划(curated)。与有核细胞相反,血小板不能转录染色体dna,阻止血小板转录因子介导基因调控,暗示rna池的转录后调控(图8a),可能是通过rna结合蛋白(rbp)(zimmermanandweyrich,2008.arteriosclthrombvascbiol28:s17-24)。实际上,sf2/asf-(srsf1-)rbp先前已涉及在健康个体血小板中启动组织因子mrna剪接(schwertzetal.,2006.jexpmed203:2433-2440)。通常,rbp涉及与基因表达相关的多个共转录和转录后过程(诸如rna剪接、多腺苷酸化、稳定化和定位)(glisovicetal.,2008.febsletters582:1977-1986)。多个rbp与rna分子的共同组装产生异质核核糖核蛋白(hnrnp),其可以定义前mrna分子的命运。5′-和3′-utr被认为是前mrna的最突出的调节区域(rayetal.,2013.nature499172-177),而内含子区域主要介导可变剪接事件(诸如外显子跳跃)。血小板rna裂解物的sage分析显示血小板含有平均更长的3′-utr长度的基因(dittrichetal.,2006.thrombhaemostasis95:643-651)。因此,我们假设rbp与血小板rna的utr区域的差异结合可以解释在tep中观察到的差异化剪接模式。我们开发了一种算法,该算法扫描utr区域中的rbp结合基序,并鉴定结合位点数量与特定基因的对数倍数变化之间的相关性。我们将该算法称为rbp-thrombo搜索引擎(图8b,参见实施例1中的详细说明)。我们包括了先前已鉴定出结合基序的102个rbp(rayetal.,2013.nature499:172-177)。我们仅包括在rna-seq数据中具有足够读数覆盖率的utr区域(图8c,参见实施例1)。我们首先鉴定了具有对5′-utr或3′-utr的富集趋向性的rbp,并且确实观察到rbm8a、fus和pprc1主要靶向5′-utr,而igf2bp2、zc3h14和raly显示对3′-utr的富集结合库(图8d)。之前报道了这些富集(rayetal.,2013.nature499:172-177),支持了我们的匹配方法的特异性。所有utr都具有用于其中一个rbp的至少一个结合位点。通过对3210个5′-utr区和3720个3′-utr区的分析,我们观察到每个utr区域的rbp结合位点的数目显示出双峰分布,表明对特定utr区域的特定rbp的受控调节(图8e、f)。为了评估nsclcteprna特征中的rna是否受特定rbp结合位点的共同调节,我们将基因的5′-utr或3′-utr的logfc值与对每个rbp的这些区域中的任何一个的匹配的结合侧的数量相关联。这生成5′-utr(fdr<0.01,rbm4、rbm8a、pprc1、fus、samd4a)的5个显著相关性和3′-utr的69个(fdr<0.01,前5个是pcbp1/2、srsf1、rbm28、lin28a和cpeb2,图8g)显著相关性。n个rbp结合位点和特征基因的logfc之间的显著相关性对于所有显著富集的rbp是正的,表明增强的结合位点可能导致增强的剪接。可能地,一经血小板活化,rbp从特定颗粒释放到血小板胞液中,从而开始剪接过程。或者,rbp由调节rbp磷酸化的蛋白激酶(诸如clk)(denisetal.,2005.cell122:379-391;schwertzetal.,2006.jexpmed203:2433-2440),和因此其细胞内定位(colwilletal.,1996.emboj15:265-275)控制。因此,我们得出结论,差异化rbp结合特征可能至少部分地促成特定tep特征,尽管需要进一步的实验验证。
实施例5
分类特征的开发
血小板在肿瘤发生和癌症转移期间充当局部和全身响应者(mcallisterandweinberg2014.naturecellbiol16:717-27),从而暴露于肿瘤介导的血小板教育,并导致血小板行为改变(labelleetal.,2011.cancercell20:576-590;schumacheretal.,2013.cancercell24:130-137;kerretal.,2013.oncogene32:4319-4324)。我们之前已经证明,通过自学习的基于支持向量机(svm)算法,血小板rna可以作为生物标志物来检测和分类来自血液的癌症(bestetal.,2015.cancercell28:666-676)(图3a)。对于血小板rna生物标志物选择和计算分析,首先对经分离的血小板rna进行smartercdna合成和扩增、truseq文库制备和illuminahiseq测序(图4d-e,实施例1)。我们将这种高度多路复用的生物标志物特征检测平台命名为thromboseq。外在因素可以影响选择过程和血小板rna生物标志物的读出(diamandis,2016.cancercell29:141-142;joosseandpantel,2015.cancercell28:552-554;fellerandlewitzky,2016.cellcommunicationandsignaling14:24),并且通过公开数据的统计建模(bestetal.,2015.cancercell28:666-676),我们能够确认个体的年龄及血液储存时间可影响血小板分类评分(表4)。因此,我们组装了来自nsclc患者(n=159)和无已知癌症患者(n=104)的血小板样品群组,匹配年龄(中位年龄(四分位差:iqr)分别为61(14.5)和58(12.25)年,图4a)和血液储存时间(血液采集12小时内血小板分离)。这个匹配的群组是更大的nsclc患者(n=352)和无已知癌症个体的群组的一部分,但不排除患有炎性疾病的个体(n=376)(图1a、表4、表5、图4a)。
匹配的nsclc/非癌症群组使我们能够首先评估潜在的技术和生物变量(即血小板活化、血小板rna产量、血小板成熟和循环dna污染)的贡献(图4-5,实施例2),以及研究血小板rna谱和rna加工途径(图1b,图5-8,实施例3-4)。此外,我们使用thromboseq平台研究了血小板rna测序效率(图4)。总之,我们的结果表明选择内含子跨接rna读取消除了血小板rna生物标志物选择过程中dna污染的潜在不良贡献,并且用于诊断算法开发的每个样品在包含之前必须检测至少3000个不同基因的库(图4)。此外,nsclc患者的剪接血小板rna谱似乎主要通过血小板教育和成熟过程中的典型剪接事件和rna结合蛋白活性的改变以响应肿瘤生长(图1b,图4-8,实施例2-4)。接下来,我们采用匹配的nsclc/非癌症血小板群组来开发nsclc诊断分类算法(图1)。我们首先通过引入基于ruv的(rissoetal.,2014.naturebiotech32:896-902)迭代校正模块改进了我们先前开发的基于svm的thromboseq分类算法(bestetal.,2015.cancercell28:666-676)的数据标准化程序的稳健性,从而显著降低相对样品间变异性(p<0.0001,双侧学生t检验,图9a-d)。其次,我们实施了pso驱动的元算法,用于选择用于分类的最有贡献的基因(图1c、图9e)。pso驱动的算法利用许多候选解决方案(即粒子),并通过采用群智能和粒子速度,该算法不断搜索更优化的解决方案,最终达到最佳拟合(kennedyetal.,2001.themorgankaufmannseriesinevolutionarycomputation.ed:davidb.fogel;bonyadiandmichalewicz2016.evolutionarycomputation:1-54)。最后,我们使用与患者年龄及血液储存时间匹配的nsclc/非癌症群组(总共n=263)测试和验证了pso驱动的thromboseq算法。我们在接收器操作特征(roc)曲线中总结了pso增强的thromboseq平台的预测测量。我们观察到这种nsclc分类算法在患者年龄及血液储存时间匹配评估(准确度:85%,auc:0.91,95%-ci:0.82-1.00,n=40,红线,图1d)和验证群组(准确度:91%,auc:0.95,95%-ci:0.91-0.99,n=130,蓝线,图1d)中具有显著的预测能力。与“匹配”评估(85%准确度)和验证群组(91%准确度)相比,训练群组的事后留一法交叉验证(loocv)分析表明性能降低(准确度:77%,auc为0.84,95%-ci:0.75-0.92,n=93,灰色虚线,图1d)。这可以通过所使用的不同分类技术来解释,并且以训练群组中的分类能力为代价优化基因小组朝向评估群组。在群增强的基因小组选择之后,训练、评估和验证群组的性能度量表明该算法尚未过度拟合,这是机器学习任务的常见缺陷(leveretal.,2016.naturemethods13:703-704)。与归因于血小板rna的预测能力相比,患者年龄及血液储存时间对癌症分类的贡献可忽略不计(表4)。值得注意的是,随机选择来自同一样品库(每组n=93)的1000名其他患者年龄及血液储存时间匹配的训练群组显示出相似的分类强度(中位auc′验证群组′:0.85,iqr:0.05),正如与随机分类相反(中位数auc′验证群组′:0.55,iqr:0.01,p<0.001)。
随后,我们涵盖完整的非匹配nsclc/非癌症群组的所有样品(分别为n=352和n=376)并开发了新的分类算法。为了开发算法训练群组,我们总结了所有匹配的患者年龄及血液储存时间样品,并分配了120个样品用于群引导的基因列表选择和svm训练,以及88个样品用于基于群的优化。因此,nsclc诊断分类器的训练群组再次没有被患者年龄或血液储存时间混淆(表4)。共有520个样品(患者年龄和/或血液储存时间不匹配),包括在多个医院和不同临床群组中收集的样品(表5),仍然用于验证算法,并且在算法的分类参数被锁定时通过算法预测。我们再次总结了在roc曲线中用于评估(准确度:91%,auc:0.93,95%-ci:0.87-0.99,n=88,红线,图1e)和验证(准确度:89%,auc:0.94,95%-ci:0.93-0.96,n=520,蓝线,图1e)pso增强的thromboseq平台的预测测量。与“完整”评估(91%准确性)和验证群组(89%准确性)相比,训练群组的事后loocv分析再次导致性能降低(准确度:84%,auc:0.90,95%-ci:0.84-0.95,n=120,灰色虚线,图1e)。在锁定基因小组的同时随机选择其他训练群组(每组n=120)引起类似的分类强度(n=1000,中位auc“验证群组”:0.89,iqr:0.05),而随机分类算法性能降低(中位auc“验证群组”:0.5,iqr:0.03,p<0.001)。因此,我们得出结论,pso驱动的thromboseq平台使得能够对基于血液的癌症诊断进行稳健的生物标志物选择,而不依赖于个体年龄、血液储存时间和某些炎性疾病引入的偏差。
实施例6响应特征的开发
接下来,我们研究了群调节tep生物标志物特征在nsclc患者治疗响应预测中的临床应用。为此,我们前瞻性地纳入了选择用pd-1单克隆抗体nivolumab治疗的nsclc患者,nivolumab在第二线设置(thesecondlinesetting)中未选择的nsclc群组中的客观响应率约为20%(borghaeietal.,2015.newenglandjmed373:1627-1639;brahmeretal.,2015.newenglandjmed373:123-135)。目前,抗pd-(l)1靶向治疗的患者的分类受到可用生物标志物的有限准确性和一致性的阻碍,包括肿瘤组织的pd-l1免疫组织化学。研究已经鉴定了肿瘤组织突变负荷、新抗原的存在,免疫细胞的浸润和对抗pd-(l)1免疫疗法的响应之间的相关性(rizvietal.,2015.science348:124-128;mcgranahanetal.,2016.science351:1463-1469)。对抗pd-(l)1免疫疗法响应可能性低的患者的鉴定,同时仍能正确识别最有可能从该疗法中获益的个体,可能会预防不必要的治疗和伴随的费用,以及患者潜在暴露于严重的免疫学不良反应事件。血小板在炎性条件下可以表现为免疫调节剂(boilardetal.,2010.science327:580-583),并因此可能还参与针对肿瘤的免疫应答。为此,我们在开始nivolumab治疗之前收集了血小板样品(n=64)。这些样品是图1a中显示的群组的一部分。用nivolumab治疗的患者的响应评估通过计算机断层扫描(ct)成像在基线、治疗开始后6-8周、3个月和6个月进行(图2a)。根据更新的实体瘤反应评估标准(recist)1.1版评估治疗响应。具有疾病控制的nsclc患者(即完全和部分响应者,以及在nivolumab治疗开始后6个月具有稳定疾病的患者)被分配到响应者组。对于thromboseq分析,我们选择了使用nivolumab治疗的64名nsclc患者的基线血液样品(n=44个响应者和n=60个非响应者),针对相对平衡的组大小,以优化开发pso驱动的nivolumab响应预测算法(图2a)。首先,我们观察到44个响应者和60个对nivolumab无响应的患者的血小板中差异化剪接rna的显著非随机聚类(通过群智能优化的基因小组,通过fisher精确检验p<0.0001,图2b)。接下来,我们重新应用了群智能来进行nivolumab响应预测特征鉴定。为此,我们随机选择了60个样品训练、21个样品依赖评估和23个样品验证的群组。使用1246个基因的nivolumab响应预测组,pso增强的thromboseq分类算法在依赖性评估群组中达到76%的准确度(auc:0.72,95%-ci:0.49-0.96,n=21,灰线,图2c)。我们接下来观察到1246个基因的nivolumab响应预测算法在独立验证群组中具有显著的预测能力(准确度:83%,auc:0.89,95%-ci:0.67-1.00,n=23,蓝线,图2c)。训练群组的事后留一法交叉验证(loocv)分析,期间60个样品训练群组的每个样品被遗漏用于算法训练并随后预测,引起高精度分类(准确度:83%,auc:0.89,95%-ci:0.81-0.97,红线,图2c)。我们通过随机选择具有相似样品大小的其他训练和依赖评估群组来确认nivolumab响应预测分类器的灵敏度(n=1000次迭代,中位数auc:0.78,iqr:0.09)。此外,我们通过在训练过程中引起随机分类的随机改类别标签(排列)确认了特异性(n=1000,中位数auc:0.30,最小-最大:0.2-0.31,p<0.0001,图2c)。使用该1246个基因分类器为nivolumab治疗(正确分配100%灵敏度)选择所有响应者的算法阈值引起53%的非响应者病例的正确分配(53%特异性,图2d)。
假设在未选择的nsclc患者群体中对nivolumab的响应率为20%(borghaeietal.,2015.newengljmed373:1627-1639;brahmeretal.,2015.newengljmed373:123-135),整个群体的42%将被安全地留在nivolumab治疗中。我们注意到在1246个基因的nivolumab响应预测算法中对n28-随访群组(在治疗开始后2-4周收集)的分类产生随机分类(数据未显示)。然而,当分别进行分析时,我们在治疗开始后2-4周观察到teprna谱中相似的独特能力(图10a),表明对于响应预测因子,在nivolumab治疗期间必须构建单独的分类器。我们还注意到,当患者用nivolumab治疗时,teprna谱改变(图10b、c)。
总而言之,我们提供了tep可能成为癌症检测和治疗选择的诊断平台的证据。pso驱动的thromboseq算法开发方法使得能够有效的选择生物标志物,并且可适用于其他诊断生物资源和适应症。通过1)对显著更多患者年龄及血液储存时间匹配样品的群增强自学习算法进行训练、2)包括小rna-seq的分析(例如mirna)、3)包括非人rna,和/或4)组合多种基于血液的生物资源(诸如teprna、外来体rna、细胞外rna和细胞外dna)可以实现群增强thromboseq的分类能力的进一步提高。从本质上讲,群智能使得能够自我重组和重新评估,从而实现连续的算法优化(图3a)。目前,对于(早期)检测nsclc和nivolumab响应预测的tep的大规模验证是有必要的。
实施例7患者情况
一名60岁的男性出现在全科医生处(gp)。他抱怨痰液混有血液、疲倦,气短、体重减轻。经过体格检查,全科医生注意到锁骨淋巴结肿大。全科医生怀疑是患有局部或转移性肺癌的患者。他下令进行基于血小板rna的诊断测试(thromboseq)。对患者进行静脉穿刺,并将全血收集在包被有edta的管中。带有血液的包被edta管通过医疗运输送到与thromboseq系统兼容的测序设施。在血液管到达测序设施后,对包被edta的管进行标准化的血小板分离方案,并从得到的血小板小球中进行总rna分离。定量、质量控制总rna,并将~500pgrna进行标准化的smartercdna扩增方案。将得到的cdna标志物用于illumina测序,并使用illumina测序平台对样品进行测序。测序后,使用thromboseq生物信息学管道处理样品的fastq文件,包括读数映射、定量、标准化和校正,并使用基于群增强的nsclcdx特征的支持向量机械(svm)分类器进行分类。分类结果将发送给gp。
一名66岁的女性被诊断出患有iv期非小细胞肺癌(nsclc),其中有多处转移到大脑。医生决定研究原发性肿瘤对抗pd(l)1靶向治疗,尤其是nivolumab治疗的敏感性。他们使用常规静脉穿刺手术抽血,并在包被有edta的真空管中收集全血。带有血液的包被edta管通过医疗运输送到与thromboseq系统兼容的测序设施。在血液管到达测序设施后,对包被edta的管进行标准化的血小板分离方案,并从得到的血小板小球中进行总rna分离。定量、质量控制总rna,并将~500pgrna进行标准化的smartercdna扩增方案。将得到的cdna标志物用于illumina测序,并使用illumina测序平台对样品进行测序。测序后,使用thromboseq生物信息学管道处理样品的fastq文件,大致包括读数映射、定量、标准化和校正,并使用基于群增强的nivolumab疗法响应特征的svm分类器进行分类。包含对nivolumab的预测响应效力的分类结果将发送给医疗团队。
实施例8最小生物标志物组
nsclc诊断基因小组
为了选择用于tep-rnansclc诊断的最小生物标志物基因小组,计算nsclc诊断评分。首先对nsclc/非癌症rna测序数据集(n=779个样品)进行ruv标准化模块(lib-大小阈值:0.418,由pso测定)。仅使用训练群组(n=120个样品)确定群组中具有稳定表达水平的基因和用于ruv校正的因子。接下来,仅使用分配给年龄、性别、edta和吸烟匹配的nsclc/非癌症训练群组的样品进行anova差异化表达分析。接下来,采用迭代生物标志物基因小组选择算法,其根据排序的fdr或p值排序的anova列表每次迭代添加新基因。生物标志物基因小组由具有正对数倍数变化的基因组成。通过选择生物标志物基因小组中基因的每个样品的中位2-log-每百万计数,每次迭代计算nsclc诊断评分。对于每个生物标志物组,评估群组(n=88)中生物标志物基因的roc曲线的auc值被评估。这是针对从2个基因到至多并包括500个基因的生物标志物基因小组进行的。
评估群组(n=88个样品)显示60个基因的生物标志物基因小组中nsclc诊断评分的roc曲线中的最高auc值(auc值:0.86,分类准确度:81%)。随后锁定60基因生物标志物基因小组和独立nsclc晚期验证群组(n=518,n=245nsclc和n=273非癌症)的roc曲线评估引起auc值为0.80(95%-ci:0.77-0.84),和分类准确度为73%,以及独立的nsclc局部先进验证群组(n=106,n=53nsclc和n=53非癌症)引起auc值为0.74(95%-ci:0.64-0.84),分类准确度为69%。
在将生物标志物基因小组减少至10个基因之前,筛选60个基因生物标志物基因小组以寻找也由pso选择的基因(参见上文)。60个基因中的45个两个基因小组中均存在,因此选择用于进一步分析。在独立的晚期验证组(n=518个样品)中,45个基因引起auc值为0.77(95%-ci:0.73-0.81)并且分类准确度为77%。在早期验证组(n=106个样品)中,auc值为0.74(95%-ci:0.65-0.83),分类准确度为70%。接下来,选择来自这45个候选生物标志物的随机10个基因小组的生物标志物基因小组(n=1000次迭代),并确定评估群组(n=88)中的分类准确性。在独立的早期和晚期验证群组中选择具有最高auc值和分类准确度(分别为0.87和81%)的随机选择的生物标志物基因小组(n=10个基因)进行验证(早期群组:n=106,auc值:0.69(95%-ci:0.59-0.79),分类准确度65%,晚期群组:n=518,auc值:0.74(95%-ci:0.70-0.77),分类准确度68%)。
用于nsclc诊断和nivolumab响应预测的p-选择素组
使用类似方法选择p-选择素5个基因特征。首先,选择与p-选择素rna的表达水平相关的所有基因,并根据相关系数和fdr值进行分类。接下来,对于在非癌症与nsclcanova中具有正的对数倍数变化的那些,筛选已分选的p-选择素相关基因。同样,根据fdr排序的p-选择素相关基因列表,通过在每次迭代中添加一个另外的基因来迭代地增加p-选择素基因小组。这是针对两个直至并包括50个基因进行的。对于每个生物标志物组,评估群组中的样品评估auc值和分类准确度,并且选择具有最佳auc值和分类准确度的p-选择素基因小组(n=5个基因,auc:0.74,分类准确度:70%)。得到的5个基因小组将独立的nsclc晚期验证样品分类,得到的auc值为0.58(95%-ci:0.53-0.62),分类准确度为57%(n=518个样品)。早期nsclc被分类为auc值为0.66(95%-ci:0.55-0.76),分类准确度为65%(n=106个样品)。
nivolumab响应预测基因小组
使用类似的方法选择用于nivolumab响应预测的最小基因小组。在治疗开始前一个月收集血小板样品(基线,n=179个样品)。在基线、治疗开始后6-8周、3个月和6个月通过ct成像进行用nivolumab治疗的患者的响应评估。根据更新的recist版本1.1标准评估治疗响应(eisenhaueretal.,2009.europjcancer45:228-247;schwartzetal.,2016.eurjcancer62:132-7),并评为进行性疾病(pd)、稳定疾病(sd)、部分反应(pr)或完全反应(cr)。主要目的是确定那些对治疗有响应的患者,而不是无响应者。因此,对于nivolumab响应预测分析,患者被分组为显示进行性疾病是非响应组中的最佳反应,总共179个样品。在任何反应评估时间点具有部分反应的患者作为最佳响应或在6个月反应评估时稳定疾病被注释为响应者,总共91个样品。为了选择和验证nivolumab生物标志物基因小组,随机选择91名响应者和91名年龄和性别匹配的无响应者,以实现相同的组大小。55名响应者和非响应者被分配到训练群组(总共n=110),25名响应者和非响应者被分配到评估群组(总共n=50),11名响应者和非响应者保持独立验证(总共n=22)。我们首先将该群组进行ruv标准化模块(jacobetal.,2016.biostatistics17:16-28)。对于该分析,选择显示与样品库大小(通过pearson相关性计算)和样品采集医院(通过anova统计计算)相关的表达水平的基因,并对样品进行ruv校正。这使得能够校正rna测序数据中的混杂因子的读数计数。仅使用训练群组确定稳定基因。接下来,我们进行m值标准化(tmm标准化;robinsonandoshlack,2010.genomebiol11:r25)的截尾均值,并对每个基因进行tmm标准化log-2转化每百万计数读数wilcoxon差异化表达分析。为此,仅包括分配至训练群组的样品。由p值分类的wilcoxon差异化表达分析得到的基因列表用作如上所述的迭代生物标志物基因小组选择算法的输入。通过从响应者中减去来自非响应者的中位计数(δ_中位值)来计算差异化表达的方向。通过从每个样品中显示表达增加的基因的中位计数减去显示表达降低的基因的中位计数来确定nivolumab响应预测得分。在迭代生物标志物基因小组选择算法的每次迭代期间,添加增加和减少的rna。对于每个生物标志物组,在评估群组中生物标志物基因的roc曲线的auc值被评估(n=50个样品)。这是针对生物标志物基因小组进行的,范围从4个直到并包括1600个基因。评估群组在4个基因的生物标志物基因小组中的nivolumab响应预测得分的roc曲线中达到最高auc值(auc值:0.69,分类准确度:70%)。随后锁定4个基因的生物标志物基因小组和独立验证群组分类的roc曲线分析(n=22,n=11个响应者、n=11个非响应者)生成的auc值为0.70(95%)-ci:0.47-0.94),分类准确度为73%。使用三种最显著的增加和三种最显著的减少的差异化表达的rna选择的6个基因的生物标志物基因小组的额外评估引起评估群组中的分类准确度为60%(auc:0.60,n=50个样品)和验证群组中的分类准确度为64%(auc:0.61,95%-ci:0.36-0.86,n=22个样品)。
- 还没有人留言评论。精彩留言会获得点赞!