使用磁阻生物传感器阵列表征DNA甲基化的制作方法
发明名称
发明领域
本申请总体涉及生物传感技术和装置。更具体地,其涉及生物传感器阵列在dna甲基化和突变分析中的用途。
背景技术:
癌症是由遗传和表观遗传改变的逐步积累引起的细胞疾病。大规模的测序工作已经确定了能够用作遗传生物标志物的复发性遗传突变,用于评估癌症发生风险,分类疾病亚型,预测对治疗的反应以及监测治疗功效。dna甲基化引起肿瘤抑制基因的表观遗传沉默,并对其在肿瘤发生中的直接影响及其作为癌症生物标志物的用途进行了研究。在膀胱和结肠癌中,遗传和表观遗传分析的组合已被证明比单独应用两种方法中的任一种具有更高的诊断价值。然而,相较于突变基因分型,甲基化表征(profiling)并不是“是-否”的结果,因为甲基化驱动的基因沉默机制通常对甲基化位点的整体密度敏感,并且基因启动子中通常存在多个cpg二核苷酸(最常见的甲基化位点)。最后,甲基化密度在单一肿瘤内的等位基因和细胞之间可能变化,从而导致异质特征。
基于扩增、探针杂交、酶消化、凝胶电泳或测序,已开发出多种技术来检测dna中的单个单点突变。dna甲基化信息在聚合酶链反应(pcr)扩增过程中丢失,并且dna杂交对靶区域的甲基化状态不敏感。因此,必须对dna进行甲基化敏感的预处理。两种主要的dna甲基化分析技术基于甲基化敏感性酶消化,亲和富集,其使用对甲基化的胞嘧啶具有特异性的抗体,或亚硫酸氢盐转化未甲基化的胞嘧啶为尿嘧啶。亚硫酸氢盐转化被最广泛地使用,因为甲基化事件被转化为单碱基改变(c/t),可以使用源自突变检测的技术进行检测,包括测序阵列杂交、甲基化敏感性pcr和甲基化敏感性解链曲线分析。对亚硫酸氢盐转化的dna进行测序将定量甲基化状态并允许比较不同测序运行和批次中的数据,但是这既昂贵又耗时。基于扩增和解链的技术对单个甲基化位点不具有特异性,并且不容易扩展以研究大量甲基化位点。基于阵列的方法,诸如illuminabeadchip(依诺米那有限公司(illuminainc.),加利福尼亚州圣迭戈)提供了高度多重化的位点特异性试验。然而,在模板的亚硫酸氢盐转化和扩增后,dna产物主要包含三个碱基(鸟嘌呤、腺嘌呤和胸腺嘧啶,加上甲基化的胞嘧啶的残基)。这种降低的序列复杂性使得用于终点检测的探针设计变得复杂,并且减少的序列变异降低了特异性。
发明概述
本发明旨在可扩展芯片平台中同时进行甲基化(和任选地,突变)表征,所述可扩展芯片平台在紧凑、易于使用且潜在的低成本平台上提供高度特异性和定量的dna甲基化和突变数据。我们的优选方法基于磁性标记的靶dna与gmr生物传感器阵列表面栓系的dna探针的杂交。为了提高dna杂交试验的特异性,我们采用了表面栓系的dna杂合体的解链曲线测量。这避免了常规试验条件优化,因为靶标-探针杂合体在解链曲线测量期间暴露于不断增加的严格性。还已经使用荧光和表面等离振子共振测量了表面栓系的dna探针的解链曲线。相较于这些方法,gmr生物传感器具有很高的灵敏度,几乎没有来自生物样品的磁性背景信号,并且不依赖于温度。
一方面,本发明提供了一种以单一位点特异性同时表征位点阵列的dna突变和甲基化事件的方法。它有利地采用以磁阻传感器阵列的甲基化检测,并同时进行dna序列中甲基化和突变的表征。使用非判别引物扩增基因组(突变)或亚硫酸氢盐处理的(甲基化)dna,然后将扩增子与磁阻(mr)生物传感器的阵列杂交,然后进行实时解链曲线测量。这种mr生物传感技术提供了可扩展的dna杂交多重检测,其已被证明对温度、ph值和生物流体基质的变化不敏感。解链曲线法进一步增强了试验特异性和对探针长度变化的耐受性。或者,该技术可以使用在阵列上施加甲基转移酶的方法,以转移拴系在传感器表面的dna序列上的甲基化位点,并直接靶向甲基化位点进行检测。该方法能够对突变和甲基化位点进行表征并提供与亚硫酸氢焦磷酸测序相当的定量评估甲基化密度。
本发明的实施方式有利地提供了可以容易地在磁性dna芯片中实施的表观遗传和突变分析。磁性检测杂交提供了高灵敏度并且几乎没有来自样品和样品基质的磁性背景。靶标-探针杂合体的实时解链曲线测量通过用越来越严格的条件挑战杂合体来提高试验的特异性。增加严格性的方法包括但不限于升高mr生物传感器阵列的温度和降低样品缓冲液中的盐(na+)浓度。重要的是,实时解链曲线测量消除了终点检测所需的探针优化需求。
一方面,一种甲基化检测的方法提供了dna链中甲基化密度的定量描述。该方法包括对含有甲基化和未甲基化位点的dna链进行亚硫酸氢盐转化,以产生具有错配碱基对的转化dna链;对转化dna链进行pcr扩增以产生pcr扩增的转化dna链;将pcr扩增的转化dna链与固定于磁阻(mr)传感器阵列上的互补dna链杂交;在杂交之前或之后,磁性标记pcr扩增的转化dna链;增加严格条件以使磁性标记的单链靶dna链由固定于磁阻(mr)传感器阵列上的互补dna链变性;在严格条件增加期间,实时读出变性的磁性标记的单链靶dna链产生的变性信号;和由变性信号确定甲基化和未甲基化dna链的严格条件。
在一个实施方式中,该方法还包括在磁性标记的单链靶dna链与固定于磁阻(mr)传感器阵列上的互补dna链杂交期间,实时读出结合信号。
严格条件可以是温度,其中增加严格条件包括在盐浓度保持恒定的同时提高温度,并且其中确定甲基化和未甲基化dna链的严格条件包括确定甲基化和未甲基化dna链的解链温度。或者,严格条件可以是盐浓度,其中增加严格条件包括在温度保持恒定的情况下降低盐浓度,并且其中确定甲基化和未甲基化dna链的严格条件包括确定甲基化和未甲基化dna的解链盐浓度。此外,严格条件可以是温度和盐的组合,其中增加严格性包括同时增加温度和降低盐浓度。
在增加严格条件的任何方法中,可以同时检测dna中的突变位点和甲基化位点。对转化dna链进行pcr扩增可包括在亚硫酸氢盐转化后和不进行转化的情况下对dna链进行pcr扩增,其中输入dna链可以包含甲基化和非甲基化位点和野生型基因和突变基因;并且由变性信号确定甲基化和未甲基化dna链的严格条件可以包括由变性信号确定甲基化和未甲基化dna链和野生型基因和突变型基因的严格条件,因此可以同时确定突变位点和甲基化位点。
因此,本发明提供了一种使用磁阻(mr)传感器阵列的甲基化检测方法。在优选的实施方式中,具有甲基化位点的dna链经亚硫酸氢盐转化并pcr扩增。然后将它们与具有固定的互补dna链的温度控制的磁阻(例如gmr)生物传感器阵列杂交并进行磁性标记。通过升高温度使靶dna链由固定的dna链变性。将来自靶dna的结合信号的实时测量用于确定解链曲线。在另一个实施方式中,使用盐浓度而不是温度来使靶dna链变性。该技术可与同一生物传感器芯片上的基因突变测量结合使用。
另一方面,一种甲基化检测的方法提供了dna序列中甲基化密度的定量描述。该方法包括对具有或不具有甲基化位点的dna链进行亚硫酸氢盐转化;pcr扩增转化dna链;转化的靶dna链与固定有(未甲基化)互补dna链的mr传感器阵列杂交;添加甲基转移酶以使对应于靶dna链甲基化位点的互补dna链甲基化;升高温度,直到靶dna链从固定的dna链变性,如果靶dna是甲基化的,则留下甲基化的单链dna,或者如果靶dna是未甲基化的,则留下未甲基化的单链dna;添加与甲基识别部分偶联的磁性纳米颗粒,如抗甲基化的赖氨酸抗体,其将结合固定于传感器的甲基化dna链;实时读取结合信号,和确定固定的dna链(以及相应的靶dna链)是否是甲基化的。
可以将不同的mr技术用于生物传感阵列。这些传感器包括但不限于巨磁阻(gmr)传感器、磁性隧道结(mtj)传感器、平面霍尔效应(phe)传感器。
附图简要说明
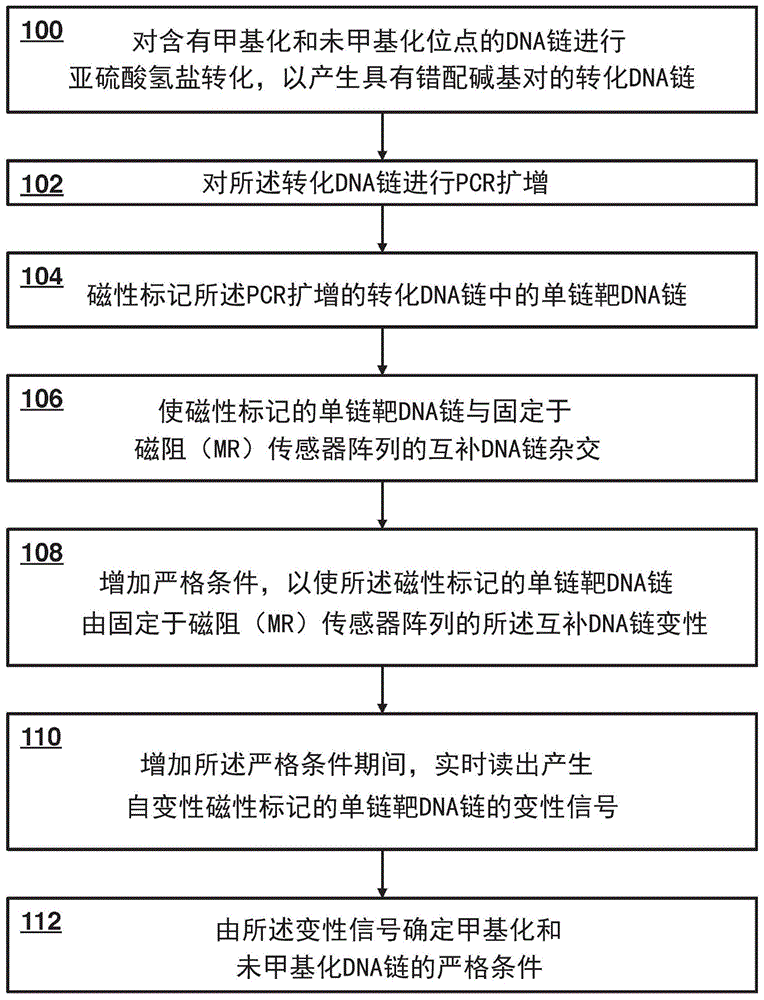
图1显示了本发明的一实施方式。
图2提供了使用gmr生物传感器装置检测磁性标记dna的示例性方案的示意图。
图3a是实时监测来自gmr生物传感器的δmr信号的图表。
图3b-c显示了靶向brafc.1391g>a突变的突变型mt探针和野生型wt探针的解链曲线。
图4a-b是亚硫酸氢盐转化过程的示意图。
图4c-d显示靶向kit甲基化(位点p1)的未甲基化(u)探针和甲基化(m)探针的解链曲线。
图5a显示了黑素瘤细胞系的突变表征。
图5b-c分别是对应图5a的热图和突变图。
图6a-c显示了黑素瘤细胞系突变和甲基化表征的结果。
图7a是差分磁阻传感器桥的示例性示意图。
图7b是温度和盐浓度解链的示意图。
图7c是示例性示意性测量设置。
图8a-b显示了与针对cd8/9突变的wt和mtdna探针杂交的分别为c(na+)=10mm和2mm的mnp标记的wtdna靶标的wt靶解链曲线。
图9a-b显示了与针对cd8/9突变的wt和mtdna探针杂交的wtdna靶标的盐浓度解链曲线。
图10显示了针对hbb的cd8/9基因座的mt探针(空心符号)和wt探针(实心符号)与wt靶标的解链温度tm和盐浓度cm的值。
图11a显示了针对cd8/9基因座以c(na+)=10mm测量的温度解链曲线。
图11b显示了针对cd8/9基因座以t=37℃测量的盐浓度解链曲线。
图11c-d显示了针对cd17基因座测量的对应温度和盐浓度解链概况。
发明详述
图1是提供根据本发明的一个实施方式的甲基化检测方法概述的流程图,所述方法提供了dna链中甲基化密度的定量描述。在步骤100中,对含有甲基化和未甲基化位点的dna链进行亚硫酸氢盐转化,以产生具有错配碱基对的转化dna链。在步骤102中,进行转化dna链的扩增。在步骤104中,pcr扩增的转化dna链中的单链靶dna链被磁性标记。在步骤106中,磁性标记的单链靶dna链与固定于磁阻(mr)传感器阵列上的互补dna链杂交。在一些实施方式中,结合信号在杂交期间实时读出。
在步骤108中,增加严格条件,以使磁性标记的单链靶dna链由固定于磁阻(mr)传感器阵列上的互补dna链变性。在步骤110中,增加严格条件期间,实时读出产生自变性磁性标记的单链靶dna链的变性信号。在步骤112中,由变性信号确定甲基化和未甲基化dna链的严格条件。
严格条件可以是温度,在这种情况下,增加严格条件包括在盐浓度保持恒定的同时提高温度。在这种情况下,确定甲基化和未甲基化dna链的严格条件包括确定甲基化和未甲基化dna链的解链温度。或者,严格条件可以是盐浓度,在这种情况下,增加严格条件包括在保持温度恒定的同时降低盐浓度。在这种情况下,确定甲基化和未甲基化dna链的严格条件包括确定甲基化和未甲基化dna链的解链盐浓度。
在一些实施方式中,该方法可以用于同时确定甲基化位点和突变位点。在这种情况下,对包含甲基化和未甲基化位点的dna链进行亚硫酸氢盐转化包括对含有甲基化和未甲基化位点的dna链以及野生型基因和突变基因进行亚硫酸氢盐转化。另外,在这种情况下,由变性信号确定甲基化和未甲基化dna链的严格条件包括从变性信号确定甲基化和未甲基化dna链与野生型基因和突变型基因的严格条件。
上述方法将通过数个实施例予以阐释。将使用下述缩写:gmr:巨磁阻;kit:酪氨酸激酶;mnp:磁性纳米颗粒;mr:磁阻;mt:突变型;pcr:聚合酶链式反应;rarb:视黄酸受体β;snp:单核苷酸多态性;wt:野生型。
dna突变分析
图2提供了根据本发明实施方式,使用gmr生物传感器装置200检测磁性标记dna的方案202的示意图。在将反向链变性并标记后,将pcr产物注入芯片204上的反应孔中。在杂交步骤212中,mnp206标记的dna与互补表面-栓系的探针于37℃杂交1小时,获得mnp208标记的杂交dna。未结合的样品通过洗涤去除。在解链步骤214中,温度从20℃扫到65℃,导致从探针210的dna变性,以测量解链温度tm。
为了检测dna突变,我们使用非判别引物pcr扩增了感兴趣的基因组区域。然后使用生物素化的引物和链霉亲和素涂覆的磁性纳米颗粒(mnp)对pcr产物进行磁性标记。磁柱分离和双链pcr产物变性后,将与mnp偶联的ssdna(mnp-ssdna)导入gmr生物传感器阵列,其中多个dna探针被分别栓系于各个传感器表面。在将注射的mnp-ssdna与表面栓系的互补探针杂交后,gmr生物传感器产生与结合的mnp成正比的传感器磁阻比变化(δmr)。为了对突变进行基因分型,我们采用了与样品的野生型(wt)和突变型(mt)序列互补的一组两个探针(补充信息表1)。在以低严格性杂交期间,扩增子以相似的亲和力与wt和mt探针杂交。为了获得单碱基特异性,通常在dna微阵列中的杂交后使用严格洗涤。为了实现更灵活的单碱基突变检测系统,我们通过提高温度并连续同时测量gmr生物传感器阵列上所有探针的dna解链来挑战杂合体。
图3a是实时监测生物传感器δmr信号的图表,所述生物传感配用正和负参照,针对brafc.1391g>a突变的突变型(mt)和野生型(wt)探针功能化。在已知wt样品与对brafc.1391g>a突变完全匹配(wt)或单碱基错配(mt)的探针杂交(37℃,1小时)期间测量信号。每条线最多对应经相同探针功能化的三个传感器。用来自est045细胞系的pcr产物进行测量,其是所研究突变的wt。在t=2分钟时注射样品。
另外,将生物素化的dna探针用作阳性参照,并将具有非特异性序列的dna探针用作阴性参照。杂交1小时后,mt探针给出比wt探针稍高的δmr比信号,这表明低严格性杂交不足以对wt样品进行基因分型。
于37℃杂交60分钟后,通过低严格性的低温洗涤除去未结合的样品。然后,以恒定速率将温度从20℃升高至65℃,同时测量解链曲线,直到所有dna杂合体解链。来自gmr生物传感器的信号(δmr)在升温期间使用传感器电阻(r)对其温度依赖性进行矫正,所述传感器电阻(r)与传感器温度线性相关。图3b和图3c显示了靶向获自指定细胞系的brafc.1391g>a突变的wt和突变型mt探针的解链曲线,其中est045和est164细胞系分别是野生型和纯合突变体。通过t=20℃的初始信号将信号标准化。解链温度tm被定义为标准化的曲线超过0.5的温度。δtm是在mt和wt探针之间解链温度的差。括号中的数字是最小有效位数的标准偏差(n=4-6)。
图3b显示了与针对c.1391g>a突变的wt和mt探针杂交的wtbraf扩增子的解链曲线。本文中,δmr信号通过t=20℃处的初始信号标准化。我们将tm解链温度定义为信号(δmr)下降到其初始信号一半(在20℃处)的温度。用两个相同的gmr生物传感器芯片重复各解链实验。三个传感器经各种探针功能化,因此各探针最多生成六个相同的解链曲线。发现所获得的解链曲线在传感器间和芯片间都是高度可重复的。图3b中靶dna与wt和mt探针的杂合体分别显示出tm(wt)=43.0(7)℃和tm(mt)=38.9(7)℃的解链温度,其中,括号中的数字是tm的标准偏差上的尾数(n≥4)。我们将解链温度差δtm定义为mt探针的解链温度和wt探针的解链温度之差δtm=tm(mt)-tm(wt)。因此,δtm<0表示靶标对wt探针的互补性比mt探针高,因此靶标是wt。所得值δtm=-4.0(3)℃与对于使用最近邻计算的wt靶标和mt探针之间的单碱基错配的期望相符28。我们还注意到,δtm的标准偏差相较于tm更低,这表明解链温度的差异比其绝对值更有重复性。
图3c显示了针对brafc.1391g>a突变杂合的细胞系测量的解链曲线22。我们发现来自wt和mt探针的解链曲线相互重叠,导致δtm=-0.6(4)℃,因为杂合样品同时包含mt和wt靶标,其与wt和mt探针杂交。wt和mt探针产生的解链曲线均由低-tm和高-tmdna杂合体所决定。因此,解链曲线重叠并呈现出较低的斜率。
dna甲基化分析
我们应用了相似的检测方案来分析靶标特定区域的甲基化状态。我们对基因组dna进行了亚硫酸氢盐处理,以将甲基化事件转化为单碱基取代(c>t)。亚硫酸氢盐转换后,我们通过非判别pcr扩增了感兴趣的基因启动子区域。
图4a、图4b是亚硫酸氢盐转化过程的示意图。亚硫酸氢盐处理402、412后,dna410中未甲基化的胞嘧啶被转化成dna414中的尿嘧啶(图4b),然而dna400中的5-甲基胞嘧啶被保留在dna404中(图4a)。在产生产物408、418的后续pcr406、416中,414中的尿嘧啶被胸腺嘧啶取代。因此,甲基化的胞嘧啶被映射到扩增子的单碱基改变(c>t)。
图4c和图4d显示靶向kit甲基化(位点p1)的未甲基化(u)探针和甲基化(m)探针的解链曲线。测量图4c超甲基化的细胞系est045和图4d未甲基化的细胞系est164的解链曲线。所述解链曲线用于评估超甲基化(est045)和野生型(est164)细胞系kit启动子(位点p1)的甲基化状态。扩增子与探针杂交,所述探针与未甲基化的(u)或甲基化的(m)靶dna互补。如先前所述测量解链曲线。本文中,将δtm定义为m探针的解链温度减去u探针的解链温度δtm=tm(m)-tm(u)。因此,负数δtm表示靶标对u探针较高的互补性和较低程度的甲基化。所研究的kit启动子约20bp区域包含三个cpg位点,其可以经甲基化(序列在补充信息表1中),并且因此我们预期比单碱基取代更高的δtm。对图4c中的超甲基化的细胞系,我们发现δtm=8.1(1)℃,这证明kit启动子的超甲基化状态,其中我们发现对于图4d中的wt细胞系,δtm=-11.7(7)℃,这表明了未甲基化状态。
黑色素瘤细胞系的多重dna表征
gmr生物传感器阵列包含64个独立传感器,他们可以分别用氨基修饰的dna探针功能化。使用上述突变和甲基化检测技术,我们一式三份同时探测braf中的3个突变位点,nras中的2个突变位点,kit启动子中的2个甲基化位点和rarb启动子中的2个甲基化位点。我们进行了7个黑素瘤细胞系的突变和甲基化表征。对于各细胞系,通过非判别pcr扩增braf和nras的靶向区域。同样,亚硫酸氢盐转化后,通过非判别pcr扩增kit和rarb的启动子区域。磁性标记后,将来自细胞系的所有扩增子的混合物注入传感器表面。对于各细胞系,使用2个名义上相同的gmr生物传感器阵列重复进行解链曲线表征。根据解链温度分析解链曲线,并且我们确定了所有研究的突变和甲基化的δtm。
图5a黑素瘤细胞系的突变表征。测量7个研究的est细胞系对于brafc.1391g>a突变的δtm。误差线是一个标准偏差(n=4-6)。水平线是用于基因分型的阈值:δtm<-2℃wt,2℃<δtm<2℃杂合mt,δtm>2℃纯合mt。图5b测量的突变和研究的est细胞系的δtm的热图。图5c测量的向基因分型突变施用阈值的δtm的热图。
图5a针对所有细胞系测量的brafc.1391g>a突变的δtm值。6个细胞系示出大约δtm=-4℃的δtm,这表明纯合wt序列。已知est164是在该位点具有杂合突变的唯一细胞系,显示出δtm=-0.5(4)℃,这与其他细胞系显著不同。
图5b的热图展示了针对各细胞系测量的所有研究的突变的δtm值。将wt(δtm<-2℃)、杂合mt(-2℃<δtm<2℃)和纯合mt(δtm>2℃)分类产生图5c所示的突变图。细胞系中鉴定的所有突变与之前的基因分型数据一致。对于细胞系est045中的nrasc.182a>t突变,我们测量到δtm=-0.2(4)℃,对于这种突变,将细胞系基因分型为杂合的;然而,已知细胞系对于该位置的a>g取代是杂合的。当采用靶向a>t突变的mt探针时,wt和mt探针与靶标相似地错配,导致δtm接近于零。tm的绝对值与其他研究的错配探针相当,确认靶标与wt和mt探针的错配。因此,可以通过来自wt探针较低的tm来检测未知突变,但试验中应当包括靶向所有可能突变的探针以进行准确的基因分型。
dna甲基化密度
甲基化表征与基因分型显著不同,因为启动子区域中的每个cpg位点的甲基化状态在等位基因之间和细胞群内各不相同。因此,将需要就样品dna甲基化部分而言进行不同的数据分析。我们使用表面-栓系的探针测量了解链曲线,所述探针靶向kit启动子的两个位置和rarb启动子的两个位置。靶向的序列包含1-4个cpg位点。将多个研究的位点与甲基化模式的固有变化结合,我们获得了分析的细胞系的δtm连续变化。图6a显示了测量的所有细胞系的δtm值。本文中,较高的δtm表示样品对m探针较高的亲和力,即超甲基化事件。复杂的δtm模式是固有甲基化变化的直接结果。
图6a-c显示了黑素瘤细胞系突变和甲基化表征的结果。图6a是对于7个研究的est细胞系测量的kit和rarb甲基化探针的δtm的热图。由于低结合信号,计算est007kit的δtm是不可能的。图6b是测量kitp1(方块)和p2(圆圈)甲基化探针位置的δtm值相对通过焦磷酸测序测量的甲基化密度的图表。图6c是测量rarbp1(方块)和p2(圆圈)甲基化探针位置的δtm值相对通过焦磷酸测序测量的甲基化水平的图表。误差线是一个标准偏差(n=4-6)。
通过亚硫酸氢盐转化的dna的焦磷酸测序独立地评估甲基化密度。对于对应探针的各靶序列,我们根据探针靶向的区域中甲基化cpg位点的数目(1-4)以及甲基化样品的比例两者来计算甲基化密度。图6b、图c显示了使用gmr生物传感器测量的δtm相对于通过针对kitp1和p2探针位置和rarbp1和p2探针位置分别进行焦磷酸测序获得的甲基化密度。在这些图中,各点对应所测量的细胞系中的一个。δtm和甲基化密度之间存在明显的线性相关性(对于所有探针位置,r2>0.94,线性回归的结果在补充表4中给出)。对于kitp1和p2探针,斜率类似(约0.22℃/%,图6b),然而对于rarb探针,对于p1和p2探针的斜率显著地不同(p1:0.076(5)℃/%、p2:0.22(2)℃/%)。p2探针的斜率是p1探针的三倍,因为p2探针覆盖了3个cpg位点,然而p1探针仅覆盖了一个cpg位点。然而,3个甲基化位点允许更复杂的甲基化位点模式,因此rarbp2探针在最佳线性拟合附近显示了更广泛的数据分布。可以对探针进行定制以牺牲线性度,以支持更高的δtm值。
这些结果证明了在gmr生物传感器上应用实时温度解链作为一种用于对甲基化密度进行表征的新型定量方法。可以使用基于阵列的方法(例如illuminabeadchips)的单碱基分辨率进行全基因组甲基化的高通量表征,但这类阵列的特异性受到亚硫酸氢盐转化的dna较低的序列变异性的限制。可以通过甲基化特异性解链曲线分析获得基因启动子的整体甲基化密度的定量值。在本文中,我们将阵列的通量和可扩展性与解链曲线分析的特异性和灵活性结合。所获得的定量表征与焦磷酸测序的结果相当。
上述示例已经说明了同时进行dna突变和甲基化表征的方法。我们的方法结合了dna微阵列技术,用于在一个平台上同时进行突变和甲基化分析。解链曲线测量用于增加突变检测的特异性。对于甲基化检测,解链曲线以与焦磷酸测序相当的水平定量甲基化状态量化。可以在能够实时监控dna杂交与温度的各种其他平台上采用相同的技术。
gmr生物传感器平台对温度的交叉敏感性低并且提供灵敏的读数。尽管它不像先进的珠微阵列系统(例如:illumina)那样提供极高的通量,但以当前的形式,gmr生物传感器平台可用于同时进行一式三份约20个突变和甲基化位点的研究。该数量对于许多临床应用而言已经足够了,这些临床应用中的重点仅限于与特定癌症相关的少量突变和甲基化位点。然而,gmr生物传感器阵列具有模块化设计,其可以扩展为包含多达数千个生物传感器27。
方法
细胞和试剂。用于该研究的黑素瘤细胞系获自欧洲可搜索肿瘤系数据库(theeuropeansearchabletumourlinedatabase,(estdab:http://www。ebi.ac.uk/ipd/estdab)),并于37℃和5%co2下保存在含有10%fbs和抗生素的rpmi-1640培养基中。用于该研究的pcr引物修饰自dahl等并获自丹麦的dna技术a/s(dnatechnologya/s)。序列可以在补充材料表2中找到。使胺修饰的dna探针(补充材料表1中的序列)与用最近邻模型计算的解链温度匹配。探针获自dna技术a/s。其他试剂:聚(乙烯-交替-马来酸酐)(西格玛-奥德里奇公司(sigmaaldrich)),聚(烯丙胺盐酸盐)(聚合科学公司(polyscience)),蒸馏水(英杰公司(invitrogen)),1-乙基-3-(3-二甲基氨基丙基)-碳二亚胺edc(西格玛-奥德里奇公司),n-羟基琥珀酰亚胺nhs(西格玛-奥德里奇公司)1%牛血清白蛋白bsa(西格玛-奥德里奇公司),磷酸盐缓冲盐水pbs(吉布可公司(gibco)),吐温20(西格玛-奥德里奇公司),尿素(费舍尔科学公司(fisherscientific)),20×盐水柠檬酸钠ssc(英杰公司),矿物油(西格玛-奥德里奇公司),mnp链霉亲和素微珠(美天旎公司(miltenyi)),磁性分离柱μ柱(美天旎公司)。
dna提取和亚硫酸氢盐处理。使用qiagenallprepdna/rna/蛋白质迷你试剂盒(凯杰有限公司(qiagengmbh),德国希尔登)分离基因组dna并用nanodropnd-1000分光光度计(nanodrop技术公司(nanodroptechnologies),特拉华州威尔明顿)进行定量。亚硫酸氢转化dna(500ng)使用ezdnamethylation-goldtm试剂盒(zymo研发公司,加利福尼亚州爱尔文)按照生产商的方案进行。
pcr扩增。进行gmr生物传感器试验之前,使用verititm96-孔热循环仪(应用生物系统公司(appliedbiosystems))和temp酶热启动聚合酶(vwr)进行pcr。这样启动所有扩增,用酶激活和dna变性,在95℃进行15分钟,40个循环的95℃进行30秒,56℃进行30秒和72℃进行30秒,然后在72℃进行10分钟的最终孵育。使用的所有引物序列可以在补充材料表2中找到。
磁性标记。如rizzi等所述处理来自各细胞系的pcr扩增的产物以获得与mnp偶联的ss-dna靶标。简言之,对于各扩增区域,将10μlpcr产物与10μl链霉亲和素包被的mnp(macs链霉亲和素微珠,目录号:130-048-102,美天旎生物技术诺登ab公司(miltenyibiotecnordenab),瑞典隆德)储液混合,并于37℃孵育30分钟。通过依次用1ml1%吐温20和1ml的0.1%bsa(含0.05%吐温20)洗涤来制备磁性分离柱(μ柱,美天旎生物技术诺登ab公司,瑞典隆德)。与磁性颗粒偶联后,将五种pcr产物混合,并在施加的磁场下添加到色谱柱中进行分离。在靶dna-珠复合物被捕获在柱中时,将反向链变性并通过在75℃下添加2ml6m尿素溶液除去。然后,去除施加的磁场,并用100μl的2xssc缓冲液洗脱偶联的复合物。
传感器制备。如之前所述,制造了带有8×8传感器阵列的gmr生物传感器芯片。芯片表面按照kim等的方法进行化学活化。简言之,将芯片依次用丙酮、甲醇和异丙醇洗涤。用氧等离子体清洗后,将芯片用聚(乙烯-交替-马来酸酐)处理5分钟。然后,用蒸馏水洗涤芯片,并使用热板在110℃烘烤1小时。用聚(烯丙胺盐酸盐)处理5分钟后,用蒸馏水洗涤芯片,并用nhs和edc的混合物活化1小时。再次用蒸馏水洗涤芯片后,使用自动阵列仪(sciflexarrayer,scienion公司)打印不同传感器上氨基修饰的dna探针(补充表2)。将各dna探针以20μm溶解于3×ssc缓冲液中,一式三份进行打印。将相同芯片上的4个传感器用生物素化的dna功能化为阳性参照,而另一组4个传感器用与任何pcr扩增的区域不互补的dna功能化为阴性参照。将芯片在室温下保存直至使用。
数据采集用于温度校正。试验之前,各gmr传感器的热电阻率针对温度校正进行描述。通过线性拟合20、30、40、50和60℃的电阻测量获得各芯片的温度系数。然后使用温度系数来跟踪各传感器的瞬时温度。
gmr生物传感器。将先前描述的测量设置用于测量gmr传感器对mnp的响应。gmr芯片的温度通过与芯片偶联的珀耳帖元件的方式进行控制。珀尔帖元件通过具有pt1000热敏电阻的lfi3751控制单元(波长电子公司(wavelengthelectronics),美国)驱动。首先,用3ml0.1%牛血清白蛋白(bsa,西格玛-奥德里奇公司)和0.05%吐温20(西格玛-奥德里奇公司)洗涤芯片。然后用100μl的1%bsa在摇床中封闭芯片1小时。封闭后,将芯片用3ml的0.1%bsa和0.05%吐温20洗涤,然后用3ml的蒸馏水洗涤。样品注射之前,在37℃进行2分钟的基线信号测量。样品注入后,在37℃测量不同传感器的dna杂交信号1小时。然后,将温度降低到20℃,以抑制样品的进一步结合并稳定dna杂合体。用100μl的0.05xssc洗涤芯片五次,以去除未结合的样品。将150μl的0.05×ssc留在反应孔中,并覆盖50μl矿物油以防止蒸发。测量所有探针的解链曲线,同时以0.05℃/s的速度将温度从20℃升至65℃。然后将温度扫回到20℃,以获得温度参照信号。
数据处理。将mnp检测为gmr传感器的磁阻效应变化(δmr)。如hall等所述使用温度参照信号计算各传感器的δmr温度系数。将5价多项式用于说明在高温度下的非线性温度依赖性。温度校正后,将20℃至65℃之间测得的解链曲线通过其在20℃的初始值进行标准化。解链温度tm被定义为标准化的信号为0.5的温度。为了计算tm,将一阶多项式拟合到感兴趣区域中的解链曲线。使用2个探针对各突变(甲基化)位点进行基因分型。将δtm定义为针对与突变的(甲基化的)序列互补的探针测量的tm减去针对与野生型(未甲基化的)序列互补的探针测量的tm之差。
焦磷酸测序。通过使用pyromarkq24平台(凯杰公司)的焦磷酸测序以后使用pyromarkq24软件的数据分析对rarb和kit启动子区域的甲基化状态进行分析。引物序列列于补充表3中。体外酶促甲基化dna(cpgenome通用甲基化dna;密理博公司(millipore))和通过全基因组扩增制备的未甲基化dna(wga;genomeplex,西格玛-奥德里奇公司)分别用作甲基化阳性和阴性对照。
表1:用于突变和甲基化表征的ssdna列表
*使用10mmna+离子浓度的最近邻(nn)模型计算理论解链温度(tm)。设计探针具有匹配的tm。
**所有探针均经过氨基标记以与gmr传感器表面结合。
表2:扩增est细胞系基因组dna的pcr引物
*kit和rarb引物被设计成扩增亚硫酸氢盐转化的启动子区域。
表3:用于对est细胞系亚硫酸氢盐转化的dna的kit和rarb启动子区域进行焦磷酸测序的引物
表4:通过焦磷酸测序的甲基化密度相对δtm的线性拟合的参数(图6a-c)。括号中的数字是拟合程序最后位数的标准偏差。
如上,我们描述了使用所谓的平面霍尔效应桥(pheb)传感器用于实时测量dna杂合体的温度解链。现在,我们描述与磁阻传感器上二维盐和温度dna变性分析有关的实施方式的示例。具体而言,这些示例说明了温度和盐浓度的组合效应并展示了靶标到wt和mt探针的二维盐和温度变性映射,以研究盐浓度解链并鉴定用于区分匹配和错配靶标-探针杂合体的最佳条件。我们进一步研究了使用单个传感器桥来区分这些靶标的wt、mt和混合物。
传感器制造
如先前所述制造磁阻传感器桥。简言之,标称组合物ta(5nm)/ni80fe20(30nm)/mn80ir20(10nm)/ta(5nm)的磁阻元件在饱和磁场中溅射沉积。ti(10nm)/pt(100nm)/au(100nm)/ti(10nm)的电接触通过电子束蒸发沉积。用900nm厚的钝化层(ormocomp,microresist技术有限公司(microresisttechnology,gmbh),德国)旋转涂覆传感器。
图7a是是根据本发明的实施方式的差分磁阻传感器桥的示意图。磁性材料是连接电接触的对角线。vx是传感器偏置电压,和vy是传感器桥输出电压。该芯片具有5个差分磁阻传感器桥,各自具有长度l=250μm和宽度w=25μm的4个传感器元件。这种传感器设计(称之为差分平面霍尔效应电桥,dpheb)测量桥的顶部两个分支与底部两个分支之间的差分信号,如rizzi等所述。
图7b是温度和盐浓度解链的示意图。升高温度(底部)或降低盐浓度(顶部)将增加严格性,从而导致靶标-探针杂合体变性。变性后,mnp标记的靶标从传感器表面释放,导致传感器桥的信号减少。
测量平台
使用商用音频扩增器708将频率为f=167hz的vrms=1.6v电压施加到并联的所有传感器桥。使用具有sr552前置扩增器的sr830锁定扩增器712(斯坦福研究系统有限公司(stanfordresearchsystems,inc.),美国)测量各传感器桥的输出电压。由于施加的偏置电流通过传感器,mnp被磁场磁化,并在第二谐波锁定信号的虚部检测到传感器上存在mnp。提供显微镜704用于对芯片的表面进行成像。
传感器芯片安装在具有良好热接触的铝芯片支架中。用pt1000温度计测量芯片座的温度,并通过驱动珀耳帖元件的lfi3751控制单元(波长电子公司,美国)进行控制。珀耳帖元件的另一侧使用商用cpu水冷却系统706冷却。
通过t分支连接到一个芯片入口的两个注射泵700、702(型号540060,tse系统公司(tsesystems),德国)在洗涤过程中在芯片中提供液体流动。通过定制的labview程序对其进行控制,以便可以在保持恒定的总液体流速的同时注入任何比例的两种液体。
传感器功能化
如rizzi等所述,可以用氨基修饰的ssdna探针选择性地功能化5个传感器桥中任一个上的传感器元件。对人β球蛋白(hbb)基因(补充信息中的序列)中snp基因分型的探针改造自petersen等并购自丹麦的dna技术a/s公司(dnatechnologya/s)。将各芯片上的传感器桥之一用作阳性参照,并将其上半部分使用生物素化的dna探针功能化。将2个传感器桥用于直接检测hbb基因cd8/9基因座的野生型(wt)或突变型(mt)变体,并将其上半部分用对应的相应探针功能化。我们将它们分别称之为mt和wt传感器。为了对hbb基因的cd8/9和cd17基因座进行wt-mt差分检测,分别使用匹配wt和mt变体的探针对芯片上的2个传感器桥的上半部和下半部功能化。
杂交。靶标标记的mnp溶液通过将10nm1:1v:v的4×盐水柠檬酸钠(ssc,吉布可公司,usa)缓冲液中的生物素化靶dna(序列在补充信息中)与美天旎链霉亲和素微珠(美天旎生物技术诺登ab公司,瑞典)储液混合至5nm的最终目标浓度c(na+)=400mm)。将靶标-mnp溶液注射到传感器,并在t=37℃下孵育30分钟。
温度解链。wtdna靶标杂交后,用稀释的ssc缓冲液在t=20℃以30μl/分钟洗涤芯片80秒至最终浓度c(na+)=10mm或2mm。洗涤后,将温度以0.1℃/s由t=20℃升高至70℃。如先前所述完全变性杂合体后,使用由t=70℃至20℃的参照扫掠,针对传感器敏感性的温度依赖性校正解链数据。
盐浓度解链。wtdna靶标杂交后,在t=30℃或40℃用2×ssc(c(na+)=400mm)以30μl/分钟洗涤芯片80秒。初次洗涤后,通过将来自注射器泵一的2×ssc缓冲液流和来自注射器泵二的milliq水混合,洗涤缓冲液浓度在1200秒内从c(na+)=400mm指数变化到0.4mm。将由没有mnp的参照浓度概况测量中确定的浓度依赖性传感器偏移量从数据中减去。
wt-mt差分测量。在针对wt-mt差分检测wt、mt和1:1wt:mt功能化的传感器桥上分析3个5nm靶dna溶液。杂交后,在温度和盐浓度解链期间测量解链曲线。如上所述用c(na+)=10mm进行温度解链。如上所述在t=37℃进行盐浓度解链。温度解链期间测量的信号针对传感器敏感性的温度依赖性进行校正并通过来自用生物素化的dna功能化的阳性参照传感器的信号标准化。
温度解链
图8a、图8b显示了与针对cd8/9突变的wt和mtdna探针杂交的分别为c(na+)=10mm和2mm的mnp标记的wtdna靶标测量的wt靶解链曲线。实时数据针对传感器输出的温度依赖性进行了校正,并标准化至20℃的初始值。如插图所示将2个传感器桥功能化。对于图8ac(na+)=10mm和图8bc(na+)=2mm,通过将温度以0.1℃/s由t=20℃升高至70℃对dna杂合体进行变性。示出一式三份的实验结果。
2×ssc缓冲液(c(na+)=400mm)中经链霉亲和素mnp标记的生物素化的wtdna靶标以dna浓度c=5nm在传感器上以t=37℃孵育30分钟。下述测量使用wt和mt传感器进行,用于snp检测hbb中的cd8/9基因座(参见图8b中的插图)。在响应所述桥的ac偏压的第二谐波桥电压(本文将其写为v)的虚部测量由于mnp的信号。当桥的上半部和下半部经历相同的mnp浓度和分布时,桥平衡并且检测到标称零信号。将mnp标记的dna与传感器桥的上半部分杂交增加了传感器桥这一半部分的局部mnp浓度并导致了v增加。
杂交后,芯片温度降低至t=20℃,以稳定杂合体并抑制进一步的结合。未结合的靶标-mnp通过洗涤去除。测试了两种缓冲液,分别具有c(na+)=10mm和2mm。洗涤后,将缓冲液停滞在传感器上,并将温度以0.1℃/s从t=20℃升至70℃,以实时测量dna杂合体的解链。
对于c(na+)=10mm(图8a),wt和mt传感器的信号稳定在20℃和30℃之间。t>30℃时,mt传感器的信号急剧下降,这表明dna杂合体的解链温度。对于wt传感器,解链偏移至更高的温度。在3个实验之间观测绝对解链温度的一些变化。然而,在各实验中,我们发现mt和wt传感器之间的温度偏移是可以重复的并且其值为约8℃。
对于c(na+)=2mm(图8b),结果遵循相似的趋势。mt传感器信号在一定温度时显示出急剧下降,该温度比wt信号低约9℃。相较于图8a,解链还在较低温度发生。
盐浓度解链
图9a、图9b显示了与针对cd8/9突变的wt和mtdna探针杂交的wtdna靶标的盐浓度解链曲线。如插图所示将pheb传感器功能化。通过在1200秒内t=30℃(图9a)和t=40℃(图9b)下将盐浓度由c(na+)=400mm降至0.4mm来使杂合体变性。示出一式三份的实验结果。
在固定温度随洗涤缓冲液中盐浓度降低变化下进行mnp标记的wtdna杂合体的解链测量。杂交后,将传感器温度设置在30℃或40℃,并用2×ssc缓冲液(c(na+)=400mm)以30μl/分钟的流速洗脱未结合的标记物。随后,洗涤缓冲液盐浓度在1200秒内由c(na+)=400mm指数降低至0.4mm,同时通过改变分别装载有2×ssc和milliq水的2个注射器泵的相对流速保持30μl/分钟的恒定总流速。在降低浓度概况期间实时测量盐浓度的解链曲线。结果示于图9a、图9b,于t=30℃和40℃处获得;因为缓冲液浓度的指数时间概况,使用对数时间标度。减去c(na+)=0.4mm处传感器信号的最终值以获得最终变化δv(c)。
在t=30℃(图9a),wt和mt传感器的信号在高盐浓度c(na+)>20mm时大致恒定,而wt信号比来自mt传感器的信号高25%。3个实验具有很高的可重复性,因此没有对数据进行标准化。mt传感器信号在c(na+)=20mm和2mm之间急剧下降,表明dna杂合体的解链。在较低的盐浓度c(na+)<3mm时,来自wt探针的信号降低。
在t=40℃时(图9b),观察到了相同的趋势,但是解链曲线偏移到了更高的盐浓度,因此与t=30℃相比,解链在更高的c(na+)下发生。
解链温度tm和浓度cm
进行误差函数与图8a,图8b中的温度解链曲线拟合以提取定义为曲线达到其初始值50%的温度的解链温度tm。对于c(na+)=10mm,我们分别针对wt和mt传感器获得了tm(wt)=43±1℃和tm(mt)=35±1℃(不确定性表明平均值(sdom)的标准误差,n=3)。c(na+)=2mm的对应值是tm(wt)=38±1℃、tm(mt)=29±1℃。
相似地,进行误差函数与图9a,图9b中的数据相对log(c)拟合以提取解链浓度cm,其被定义为误差函数达到其初始值50%的点。在t=30℃处,我们获得了cm(wt)=1.4±0.1mm和cm(mt)=6.3±0.3mm,并且在t=40℃处,我们获得了cm(wt)=5.7±0.2mm和cm(mt)=15±1mm。不确定性表明sdom(n=3)。
图10显示了针对hbb的cd8/9基因座的mt探针(空心符号)和wt探针(实心符号)与wt靶标,获自分别与温度和盐解链拟合的误差函数的解链温度tm和盐浓度cm的值。虚线表示所用温度(水平)和浓度(垂直)概况。箭头表示增加严格性的方向。误差线是平均值的标准偏差(n=3)。获自变性实验的tm和cm的值收集在图10中。虚线表示所用的温度或浓度概况。相较于错配的mt探针-wt靶标杂合体,完美匹配的wt探针-wt靶标在变性点具有更高的严格性。两者之间的间隔不是恒定的,但在图的中心区域最大。
使用wt-mt差分测量的基因分型
先前我们已经证明,当单个传感器桥的上半部分和下半部分分别用wt和mt探针功能化时,其可以将其用于基因分型。传感器输出与绑定到传感器桥上半部分和下半部分的mnp量中的差异成比例。为了对hbb基因的各cd8/9和cd17基因座进行相对于表1所示突变的基因分型,我们用wt和mt探针分别在传感器桥上半部和下半部分进行功能化(参见图11a-d中的插图)。测量了三个靶标组合:wt、mt和1:1wt:mt。
图11a显示了针对cd8/9基因座以c(na+)=10mm测量的温度解链曲线。注意的是,曲线显示了相对于获自阳性参照传感器的信号的信号。在低温下,所有3个靶标的相对信号均接近于零,这表明靶标与wt和mt探针的杂交相同。在温度t=30℃至50℃时,来自3个靶标的相对信号彼此明显不同。对于wt靶标,相对信号的峰值为正值0.17,而对于mt目标,信号的峰值为负值-0.15。来自1:1wt:mt靶标混合物的信号保持接近零的值并达到-0.06的最小值。在t=50℃以上时,3个靶标的相对信号稳定在零。
图11b显示了针对cd8/9基因座以t=37℃测量的盐浓度解链曲线。在高盐浓度(c(na+)>50nm)时,wt和mt靶标在范围δv=-0.005μv-0μv显示出相似的值。来自混合的wt:mt靶标的信号显示较低的值(δv=-0.021μv),随着c(na+)增加该值趋向于零。对于c(na+)=40nm-c(na+)=4nm,三个靶标显示出最大的分离,其中wt靶标达到δv=+0.023μv,mt靶标达到δv=-0.023μv,而混合的mt:wt靶标显示出接近δv=0μv的稳定信号。在低盐浓度(c(na+)<4nm)时,来自三个靶标的信号稳定在零。
图11c、图11d显示了针对cd17基因座测量的对应温度和盐浓度解链概况。在温度解链研究中(图11c),三个靶标即使在低温度下也显示出清晰的分离。温度的这种趋势对于cd8/9基因座也相同,除了峰出现在较低的温度且峰水平略低。观测到三个靶标之间的最大分离在35℃和40℃之间。在盐浓度解链研究中(图11d),三个靶标最初以高c(na+)分离,δv(wt)>δv(wt:mt)>δv(mt)>0。增加严格性(降低c(na+))后,对于所有三个靶标的δv下降并彼此分离。在c(na+)=29mm观察到最大的分离并且对应δv(mt)=+0.015μv、δv(mt)=-0.008μv和δv(wt:mt)=+0.002μv。对于c(na+)<7mm,所有靶标的信号趋于δv=0。
解链曲线讨论
桥式传感器的差异设计允许实时测量dna杂合体的形成和解链。将微流体系统中的传感器芯片与温度控制集成使得我们能够进行作为与温度和/或盐浓度的函数的二维解链研究。这两个参数都将影响dna杂合体的稳定性,因此可用于区分完美匹配的靶标-探针杂合体和低至单核苷酸水平的错配。该磁性传感器平台结构紧凑并且足够稳健,能够在不同的温度或盐浓度条件下进行读取。测量温度和盐浓度解链曲线以描述hbb基因中研究的基因座。在两个研究中,相较于完美匹配的对应物,不匹配的mt探针-wt目标双链体在较低的严格性(较低的t,较高的c(na+))下变性。正如最近的邻居模型所预期的那样,降低缓冲液c(na+)导致匹配和错配的杂合体较低的tm。相似地,增加t将导致较高的cm。
图11a-d不同传感器针对靶dna的wt、mt和1:1wt:mt混合物测量的解链曲线(图11a、图11c)和盐浓度变性曲线(图11b、图11d)。用针对cd8/9基因座(图11a、b)或cd17基因座(图11c、图11d)的wt和mt探针功能化传感器,如插图所示。
两种方法在可重复性上显示出明显的差异。尽管盐浓度变性曲线(图9a、图9b)几乎完全重叠,但为重复测量而测得的绝对解链温度却显示出约2℃的不确定性(sdom,n=3)。针对两个传感器同时测量的解链温度的差异δtm=tm(wt)-tm(mt)为δtm=7.7±0.2℃(c(na+)=10mm)和8.9±0.5℃(c(na+)=2mm),其中所述的不确定性为sdom(n=3)。因此,δtm的不确定性明显低于绝对解链温度的不确定性。这表明很难准确复制相同的温度概况。例如,绝对温度的变化可能源自洗涤缓冲液或芯片周围的温度的不同。我们的研究结果进一步表明,盐浓度概况更容易被复制。此外,来自磁阻传感器的信号对盐浓度中的变化仅具有弱敏感性。此外,温度控制的要求对盐浓度解链的限制较小,并且可以使用简单的两泵设置来改变浓度。因此,浓度解链实验更容易实现。
tm和cm的二维映射
解链曲线随温度和盐浓度变化变化的测量结果使得我们在实验条件的这两个维度上能够映射变性点。产生的图10的映射图表示测量的tm和cm。这些结果提供了对(t,c(na+))-平面中各种严格性下dna杂合体的稳定性提供了重要的了解。
基于杂交的试验旨在区分匹配和错配的探针-靶标杂合体。在端点检测方案中,这是通过选择严格条件来完成的,该条件会尽可能使错配的杂合体变性并维持匹配的杂合体。同样,变性试验应当使匹配的和错配的杂合体的熔点之间的差距最大化。在图10中,我们可以鉴定介于t=32-38℃和c(na+)=3-7mm之间的最佳区域,其中空心符号和实心符号之间的距离最大。
一方面,所述磁阻传感器检测方案和设置允许(t,c(na+))-平面中的任何概况,即针对t和c(na+)的同时改变。图10表明wt和mt之间潜在更好的分离可以通过同时升高温度并降低盐浓度沿着(t,c(na+))-平面中的对角线获得。这将是未来研究的主题。
文献中已经提出了具有多达数千个传感器的磁阻传感器阵列16。与wt和mt检测探针的阵列杂交的dna靶标解链的实时二维图谱能够高度平行地筛选一系列序列和探针长度的条件,以靶向dna靶标中的多个基因座和基因。这可以用于直接基因分型,但是也可以用于鉴定(t,c(na+))-平面上经严格洗涤后最适合端点检测的区域。这可以显著地简化微阵列的设计并改善其性能,其中探针的设计可能具有挑战性,因为单次严格洗涤必须在匹配和错配的杂合体之间产生很大的差异,同时仍要保持来自匹配杂合体的显著信号。
使用wt-mt差分测量的基因分型
通过使用wt和mt探针对单个传感器桥的上半部分和下半部分进行功能化,有可能测量靶标与两种探针的差异结合。对于给定的传感器阵列,该配置允许研究更多数量的突变位点,因为对于各突变仅使用一个传感器。
图11a、图11b的解链曲线显示了cd8/9和cd17基因座不同的表现。靶标-探针杂合体的稳定性取决于探针的长度,其c+g含量和所研究的突变类型。本文中,cd8/9基因座处的突变是单碱基(c)插入,而cd17基因座处的突变是单碱基(t>a)转化。探针对于cd8/9和cd17基因座的长度分别为22个碱基(c+g54%)和18个碱基(c+g66%)。
cd47较短的探针长度导致三个靶标也在低严格性分离(图11c、图11d)并且在较低的严格性下发现了靶标之间的最大分离,相较于cd8/9。而且,cd8/9中的碱基插入导致更不稳定的错配杂合体,并导致三个靶标之间更高的分离(图11a、图11b)。
对于cd8/9基因座,来自图5b中以高c(na+)混合的1:1wt:mt的初始负信号可能是由于mt靶标-mt探针杂合体相较于wt靶标-mt探针较高的亲和力。这受到这样观察的支持,即来自mt靶标的信号以比图11b中wt靶标更低的c(na+)(较高的严格性)达到峰值。
对于cd17基因座的盐浓度解链(图11d),来自所有三个靶标的初始信号在高c(na+)下都是正信号,并发现随着盐浓度的降低而降低。我们推测这是由于与mt探针相比,与wt探针的更高的非特异性结合引起的。
wt和mt探针对cd8/9和cd17基因座的不同表现将需要在终点检测中优化测定方法,以确定最佳的洗涤严格性,以进行正确的基因分型。相反,使用变性曲线方法,杂合体会受到不断变化的严格性的限制,并且因此我们可以轻松地区分三种不同的靶标组成。
结论
上述示例证明了集成在芯片实验室(lab-on-a-chip)系统中磁阻传感器阵列在研究dna杂合体变性随温度和盐浓度变化中的应用。磁性读数对变化的实验条件仅具有较弱敏感性,因此可以用于提供灵敏的信号实时读数,所述信号来自与检测探针杂交的磁性纳米颗粒标记的dna靶标。差分传感器设计能够研究wt靶标与针对人hbb基因的两个基因座的wt和mt检测探针的特异性结合。在温度-盐浓度平面中不同切口进行的解链实验鉴定了两者之间的最佳区分区域。此外,我们发现盐浓度解链曲线比温度解链曲线更可再现。这些为芯片实验室系统中的温度解链曲线提供了迄今为止尚未研究但有趣的替代方法。
此外,这些示例还证明了使用单个传感器桥区分wt、mt和1:1wt:mt靶标,所述单个传感器桥在其顶部和底部分别经wt和mt探针功能化。对温度解链和盐浓度解链进行该操作。
这证明了使用芯片实验室磁阻传感器阵列描述dna杂合体稳定性随盐浓度和温度变化的可行性。在较大的传感器阵列上,这可用于同时映射温度-盐浓度平面中多个探针-靶标相互作用,用于实时检测或鉴定最佳测定条件的区域。
- 还没有人留言评论。精彩留言会获得点赞!
- 检测dna甲基化的方法和试剂盒的制作方法
- 可用于检测与2型糖尿病相关的ptpn1基因启动子区甲基化程度的试剂盒及其应用
- 可用于检测与2型糖尿病相关的glud1基因启动子区甲基化程度的试剂盒及其应用
- 一种基于量子点荧光共振能量转移检测基因甲基化程度的方法
- 可用于检测与大肠癌相关的mdfi基因启动子区甲基化程度的试剂盒及其应用
- 可用于检测与大肠癌相关的PCDH-γ-A12基因启动子区甲基化程度的试剂盒及其应用
- 可用于检测与大肠癌相关的SLCl9A1基因启动子区甲基化程度的试剂盒及其应用
- 可用于检测与大肠癌相关的sstr2基因启动子区甲基化程度的试剂盒及其应用
- 可用于检测与大肠癌相关的cmtm3基因启动子区甲基化程度的试剂盒及其应用
- 基因甲基化在调控基因表达方面的应用