用于修饰靶序列的多核苷酸及其用途的制作方法
本发明涉及以基因治疗、品种改良、生物技术创造等为目的而对基因序列进行精密修饰的分子遗传学技术等。
背景技术:
作为以基因修饰为目的通过切割作为标靶的基因组的碱基序列位点来提高基因修饰频率的技术,已知有zfn法、talen法、crispr/cas9法(morton,j.,etal.,proc.natl.acad.sci.usa103,16370-16375(2006);cermak,t.etal.,nucleicacidsres.39,e82(2011);cong,l.etal.,science339,819-823(2013);mali,p.etal.,science339,823-826(2013))。
然而,即使想要导入修饰碱基序列的供体dna、并通过上述3种方法中的任一种来修饰靶位点,也难以获得靶位点被准确地转化为想要导入的修饰碱基序列而不包含除此以外的修饰的细胞。
例如,在zfn法、talen法、crispr/cas9法中,极其严格地控制靶序列的特异性是很困难的,并且切割与靶序列类似的脱靶序列,因此,有时在再结合时产生被称为indel(插入缺失标记)的插入或缺失(不正确的再结合)(off-target不正确再结合问题)。另外,即使切割靶序列,也存在在再结合时导入indel的隐患(on-target不正确再结合问题)。但是,在基因治疗等精密基因修饰时,必须避免这些现象。
另外,为了选择目标修饰序列被导入基因组的细胞,通常可以使用药物选择标记基因。在现有的方法中,构建在与想要修饰的外显子的碱基序列邻接的内含子的碱基序列中插入了该标记基因的载体,将该载体导入细胞,诱导同源重组,然后进行药物选择。但是,由于得到的细胞当然具有标记基因,因此,在该情况下,该标记基因残留在细胞的基因组中引入的内含子的碱基序列中。
为了去除该标记基因,可以使用如下供体dna,所述供体dna在位于与作为目标的外显子修饰碱基序列紧邻的内含子中插入了被位点特异性重组酶识别序列(loxp、frt)夹住的药物抗性标记基因。将该供体dna导入细胞,切割该外显子-内含子区域内,分离出将从内含子药物抗性标记基因至外显子修饰碱基序列的整个长链引入至基因组中的细胞克隆,然后对该细胞克隆导入位点特异性重组酶(cre,flp)基因,获得仅去除了药物抗性标记的细胞(li,h.l.etal.,stemcellreports4,143-154(2015))。
根据该方法,可以通过位点特异性重组仅将被位点特异性重组酶识别序列(loxp,frt)夹住的选择标记基因去除,但在以cre/loxp法、flp/frt法为代表的方法中,由于重组酶的特性,一个识别序列未被去除而残留下来,因此存在无法适用于基因治疗、精密的基因修饰的问题(位点特异性重组序列的残留问题)。
另外,在该方法中,在选择导入了供体dna的细胞之后,需要将表达位点特异性重组酶的载体等导入细胞的追加工序,进一步还需要对于将导入的位点特异性重组酶表达载体从细胞中去除进行确认等,步骤繁杂。
在其它方法中,使用如下供体dna,所述供体dna在位于与作为目标的外显子修饰碱基序列紧邻的内含子中插入了被转座酶识别序列(piggybacitr)夹住的药物抗性标记基因。将该供体dna导入细胞,切割该外显子-内含子区域,分离出将从内含子药物抗性标记基因至外显子修饰碱基序列的整个长链引入了基因组的细胞克隆,然后,对该细胞克隆导入转座酶(piggybactransposase)基因,获得去除了药物抗性标记和识别序列(piggybacitr)的细胞(yusa,k.etal.,nature478,391-394(2012))。
然而,该方法存在2个问题。一个问题是被piggybacitr夹住的选择标记基因的插入位置限定于ttaa序列,换言之,如果在修饰对象位点附近不存在ttaa序列,则无法应用;另一个问题是即使可以应用,在选择导入了供体dna的细胞之后,也需要将表达转座酶的载体等导入细胞的追加工序,进一步还需要对于将导入的转座酶表达载体从细胞中去除进行确认等,步骤繁杂。
如上所述,在使用zfn、talen、crispr/cas9的方法中,特异性切割进行基因组编辑的对象细胞的基因组的靶位点,并在该位点与供体质粒中的靶序列发生同源重组,但在依赖于该机制的方法中,不仅发生染色体基因座上的靶序列的切割,而且发生来自供体质粒的靶序列的切割时,同源重反应受到抑制,因此需要通过去除供体质粒上的靶碱基序列(或者转变为非靶序列)来避免来自供体质粒的靶序列的切割。然而,如果为此而修饰供体dna的序列,则在基因组中引入了该序列的细胞中,基因组原本的序列从该区域失去,因此存在无法应用于基因治疗、精密的基因修饰的问题(失去切割位点的基因组序列的问题)。
现有技术文献
非专利文献
非专利文献1:morton,j.,davis,m.w.,jorgensen,e.m.&carroll,d.inductionandrepairofzinc-fingernuclease-targeteddouble-strandbreaksincaenorhabditiseleganssomaticcells.proc.natl.acad.sci.usa103,16370-16375(2006)
非专利文献2:cermak,t.etal.efficientdesignandassemblyofcustomtalenandothertaleffector-basedconstructsfordnatargeting.nucleicacidsres.39,e82(2011)
非专利文献3:cong,l.etal.multiplexgenomeengineeringusingcrispr/cassystems.science339,819-823(2013)
非专利文献4:mali,p.etal.rna-guidedhumangenomeengineeringviacas9.science339,823-826(2013)
非专利文献5:li,h.l.etal.precisecorrectionofthedystrophingeneinduchennemusculardystrophypatientinducedpluripotentstemcellsbytalenandcrispr-cas9.stemcellreports4,143-154(2015)
非专利文献6:yusa,k.etal.targetedgenecorrectionofα1-antitrypsindeficiencyininducedpluripotentstemcells.nature478,391-394(2012)
技术实现要素:
发明要解决的课题
本发明涉及用于高效地修饰基因组序列的供体多核苷酸、以及使用了该供体多核苷酸的基因组修饰细胞的制造方法等。
解决课题的方法
on/off-target不正确再结合
如上所述,由于目前基因组编辑所用的酶是切割染色体的靶序列的酶,因此存在以下隐患:发生切割标靶以外的染色体位点的off-target(脱靶)的风险、修复被切割的基因组时导入indel的风险。为了避免该问题,本发明人等考虑仅切割供体质粒而不是切割染色体。例如,如果在导入前切割包含供体质粒的修饰后序列的同源区域并将直链型供体质粒导入细胞、或者在将环状的供体质粒导入细胞后在细胞内序列特异性地切割供体质粒,则不会发生靶序列或与靶序列类似的脱靶序列(on/off-target)的切割,因此不会发生不正确的再结合,因此不会导入indel,成为与基因治疗等精密基因修饰相适应的技术。
位点特异性重组位点残留
另外,为了在基因组编辑中仅向靶位点导入必要的修饰而不残留标记基因、除去标记基因的痕迹,本发明人等考虑在供体质粒中的基因组片段的外侧配置阳性选择标记基因和阴性选择标记基因两者。在该情况下,由于供体质粒的骨架内配置有2个选择标记基因,因此,通过在靶序列区域发生的同源重组而插入整个供体质粒,形成来自供体质粒的修饰后的碱基序列和染色体上的修饰前的碱基序列(无特定顺序)夹住供体质粒的骨架区域且串联排列的载体靶向插入体结构(vector-insertedtargetconstruct)。由于该中间体结构中包含的修饰后碱基序列和修饰前碱基序列容易发生同源重组,因此,通过仅培养基因组中包含该结构的细胞,就可自发地置换为仅由修饰后碱基序列形成的单独结构。在该过程中不需要位点特异性重组酶、转座酶等,因此能够极其简便地获得目标细胞,而且从不存在特异性重组识别序列残留的可能性的观点考虑,也是优异的。
靶切割序列的失去
另外,如上所述,如果不仅发生染色体基因座上的靶序列的切割,还发生来自供体质粒的靶序列的切割,则供体质粒上长链区域与基因座上靶位点的同源重组反应受到抑制。在现有方法中,通过从供体质粒上去除切割序列来避免供体质粒侧的同源区域的切割,但在这样的方法中,存在去除了切割序列的修饰序列被引入细胞的染色体的隐患。与此相对,在本发明的一个方式中,通过在导入前切割包含供体质粒的修饰后序列的同源区域、并导入直链型供体质粒,从而避免染色体基因座上的靶序列和来自供体质粒的靶序列这两者的切割。另外,在本发明的另一个方式中,在供体质粒中的基因组片段中插入例如以i-scei为代表的序列特异性切割酶的识别序列,在将该供体质粒导入细胞之前、之后、或同时,使序列特异性切割酶基因在细胞内表达,切割供体质粒内的识别序列。由于序列特异性切割酶的识别序列不存在于细胞所具有的染色体的基因座上的相应区域,因此可以避免染色体基因座上的靶序列和来自供体质粒的序列的双切割,能够防止同源重组效率的降低。
这样,本发明提供解决现有的基因组编辑技术所具有的课题的新型基因组编辑技术,由此可以仅将目标修饰简便且正确地导入基因组。
即,本发明涉及用于修饰基因组序列的新型的供体多核苷酸及其用途等,更具体而言,涉及各项权利要求记载的发明。需要说明的是,2个或2个以上引用同一权利要求的权利要求所记载的发明的任意组合所形成的发明也是本说明书包含的发明。即,本发明涉及以下的发明。
〔1〕一种供体多核苷酸,其用于修饰基因组序列,包含含有1个或多个修饰的基因组片段,该基因组片段的两端通过多核苷酸连接在一起,该连接多核苷酸(linkerpolynucleotide)中包含阳性选择标记基因及阴性选择标记基因这两者,该基因组片段可切割,该可切割的位点被切断而形成供体多核苷酸链的两末端,由此,供体多核苷酸可以成为直链状,该位点也可以在供体多核苷酸中连接而使供体多核苷酸成为环状。
〔2〕根据〔1〕所述的供体多核苷酸,其中,在该可切割位点添加有切割序列。
〔3〕根据〔2〕所述的供体多核苷酸,其中,添加于该位点的切割序列不包含在与供体多核苷酸所含的基因组片段相对应的作为标靶的细胞的基因组片段的序列中。
〔4〕根据〔1〕~〔3〕中任一项所述的供体多核苷酸,其中,在该基因组片段中,该1个或多个修饰仅包含于该位点的一侧。
〔5〕根据〔1〕~〔4〕中任一项所述的供体多核苷酸,其中,该连接多核苷酸(linkerpolynucleotide)是质粒的多核苷酸。
〔6〕根据〔1〕~〔5〕中任一项所述的供体多核苷酸,其中,在阳性选择标记基因及阴性选择标记基因之间不包含作为标靶的细胞的基因组序列。
〔7〕根据〔1〕~〔6〕中任一项所述的供体多核苷酸,其中,阳性选择标记基因及阴性选择标记基因融合在一起,阳性选择标记和阴性选择标记以融合蛋白质的形式表达。
〔8〕一种修饰基因组序列的方法,该方法包括:(a)将〔1〕~〔7〕中任一项所述的供体多核苷酸导入细胞的工序、(b)通过阳性选择标记选择导入了该供体多核苷酸的细胞的工序、以及(c)通过阴性选择标记选择去除了该连接多核苷酸的细胞的工序。
〔9〕根据〔8〕所述的方法,其中,在工序(a)中包括将切断供体多核苷酸的该位点而成的直链状供体多核苷酸导入细胞的工序。
〔10〕根据〔8〕所述的方法,其中,在工序(a)中包括将连接有该位点的环状供体多核苷酸、以及切割该位点的切割酶或表达该酶的载体导入细胞的工序。
〔11〕根据〔10〕所述的方法,其中,同时导入该环状供体多核苷酸、以及该酶或该载体。
〔12〕根据〔10〕或〔11〕所述的方法,其中,表达该酶的载体是表达该酶的负链rna病毒载体。
〔13〕根据〔8〕~〔12〕中任一项所述的方法,其还包括选择基因组中包含目标修饰的细胞的工序。
〔14〕根据〔8〕~〔13〕中任一项所述的方法,其用于在遗传性疾病的致病基因中将致病序列转变为正常序列。
〔15〕一种细胞,其具有基因组中引入了〔1〕~〔7〕中任一项所述的供体多核苷酸的结构,供体多核苷酸中包含的修饰基因组片段和与其对应的来自细胞的基因组的片段具有经由该连接多核苷酸无特定顺序地串联而成的结构,在该连接多核苷酸中包含阳性选择标记基因及阴性选择标记基因。
〔16〕一种基因组经过修饰的细胞的制造方法,该方法包括:通过阴性选择标记选择〔15〕所述的细胞,选择去除了该连接多核苷酸的细胞的工序。
〔17〕一种载体,其是〔12〕所述的方法中使用的负链rna病毒载体,其编码切割供体多核苷酸的切割位点的核酸内切酶。
〔18〕根据〔17〕所述的载体,其是仙台病毒载体。
〔19〕根据〔17〕或〔18〕所述的载体,其中,该核酸内切酶为i-scei。
〔20〕用于〔12〕所述的方法的组合物,其包含〔17〕~〔19〕中任一项所述的载体。
需要说明的是,本说明书中记载的任意技术事项及其任意组合均包含于本说明书。另外,在这些发明中,除本说明书中记载的任意事项或其任意组合以外的发明也包含于本说明书。此外,关于本发明,说明书中记载的某个特定方式不仅公开了其本身,而且是包括该方式在内的更上位的本说明书中公开的发明,因此也公开除该方式以外的发明。
发明的效果
根据本发明,可以提供精密修饰基因序列的分子遗传学技术。本发明可以用于基因治疗、品种改良、生物技术创造等各个方面。
附图说明
图1是示出新型基因组编辑技术的概要的图。
图2是示出通过新型基因组编辑技术进行基因修饰的验证实验的图。
图3是示出了确认载体靶向插入体的结构的方法的图。
图4是示出了从载体靶向插入体之一通过更昔洛韦选择来分离基因修饰体的方法的图。
图5是示出了基因修饰方法:现有方法的问题点及本发明的优异性的图。
具体实施方式
本发明提供用于修饰基因组序列的新型供体多核苷酸。该多核苷酸包含含有1个或多个修饰部位的基因组片段,该基因组片段的两端通过多核苷酸连接在一起(在本发明中,将其称为连接多核苷酸),该连接多核苷酸中包含阳性选择标记基因及阴性选择标记基因这两者。
基因组片段的来源物种没有特别限制,可以是例如希望的真核生物的基因组,也可以是来自于例如酵母、动物细胞、植物细胞等的基因组片段。优选为来自动物细胞的基因组片段,更优选为哺乳动物细胞,例如灵长类的细胞,具体可以列举:小鼠、大鼠、猴及人的细胞的基因组片段。
另外,该基因组片段可以在任意位置进行切割。该可切割的位点被切断而成为供体多核苷酸链的两末端,由此,供体多核苷酸可以成为直链状(线性),该位点可以在供体多核苷酸中连接而使供体多核苷酸成为环状。在该可切割的位点被切割而使供体多核苷酸成为直链状的情况下,该多核苷酸的原本为1个的基因组片段断开而成为2个,分别结合于连接多核苷酸的两端。即,成为连接多核苷酸被一对基因组序列夹住的结构。在供体多核苷酸成为环状的情况下,基因组片段的一端与连接多核苷酸的一端连接,基因组片段的另一端与连接多核苷酸的另一端连接,成为环状的结构。
供体多核苷酸中包含的基因组片段可以包含1个或多个修饰部位。这里,修饰部位是指具有与靶细胞带有的基因组相对应的部位的序列不同的序列的部位。这些修饰部位与想要进行基因组编辑的对象细胞的基因组序列不同,通过导入该供体多核苷酸,对象细胞的基因组序列被修饰。修饰部位的数量没有特别限定,可以是1个或1个以上的部位,例如可以为2、3、4、5、10个部位、或更多部位。另外,各修饰可以是1个或多个碱基取代、碱基插入和/或碱基缺失、或者是它们的组合。
如上所述,供体多核苷酸中包含的基因组片段的两端通过多核苷酸连接在一起。连接多核苷酸与基因组片段连接成一系列连续的双链核酸。连接多核苷酸的序列没有特别限制,可以是来自例如质粒载体、噬菌体载体、粘粒载体、病毒载体、人工染色体载体(包括例如酵母人工染色体载体(yac)及细菌人工染色体载体(bac))等的序列。这样,连接多核苷酸具有载体骨架,在作为载体而发挥功能时,本发明的供体多核苷酸也称为供体载体(donorvector)。在载体为质粒载体的情况下,本发明的供体多核苷酸也称为供体质粒(donorplasmid)。作为载体而发挥功能的供体多核苷酸可以保持在适当的宿主(细胞、大肠杆菌)内。另外,在载体具有复制能力的情况下,供体多核苷酸可以在宿主内复制。本发明的供体多核苷酸优选在适当的宿主内具有自主复制能力。
连接多核苷酸中可以包含想要进行基因组编辑的对象细胞的基因组序列,如上所述,想要进行修饰的靶基因组序列仅是连接在连接多核苷酸的两端的基因组序列,连接多核苷酸中可以包含的基因组序列不是想要进行修饰的靶序列。
连接多核苷酸的长度没有特别限制,可以适宜地制成适当长度的多核苷酸。在使用载体的骨架作为连接多核苷酸的情况下,连接多核苷酸的长度可以根据载体的种类而改变。作为一个例子,连接多核苷酸的长度(包括后述的阳性选择标记基因及阴性选择标记基因的长度)例如可以为1kb以上、2kb以上、3kb以上、5kb以上、7kb以上、10kb以上、20kb以上或30kb以上。另外,可以为100kb以下、800kb以下、70kb以下、60kb以下、50kb以下、40kb以下、30kb以下、20kb以下、10kb以下或8kb以下。
在本发明中,连接多核苷酸包含阳性选择标记基因及阴性选择标记基因。这里,阳性选择标记基因是指编码用于选择保持该标记的细胞(和/或去除不具有该标记的细胞)的标记的基因,阴性选择标记基因是指编码用于去除保持该标记的细胞(和/或选择不具有该标记的细胞)的标记的基因。阳性选择标记基因及阴性选择标记基因可以适当选择。例如,作为阳性选择标记基因,可以示例出hyg(潮霉素抗性基因)、puro(嘌呤霉素抗性基因)、β-geo(β半乳糖苷酶与新霉素抗性基因的融合基因)等各种抗药性基因等,但并不限定于此。作为阴性选择标记基因,可以举出例如直接或间接诱导抑制细胞增殖或生存的基因,具体可以列举:来自单纯疱疹病毒的胸苷激酶(tk)基因、白喉毒素a片段(dt-a)基因、胞嘧啶脱氨酶(cd)基因等,但并不限定于此。
以往的基因组编辑中使用的供体多核苷酸通常在供体多核苷酸所包含的基因组片段中具有阳性选择标记基因,在配置阴性选择标记基因的情况下,配置在基因组片段的外侧。与此相对,本发明的供体多核苷酸的特征在于,在连接多核苷酸中包含阳性选择标记基因和阴性选择标记基因这两者。即,优选在阳性选择标记基因与阴性选择标记基因之间不包含靶细胞的基因组序列、或者即使包含也是不发生重组的程度的短基因组序列。这样的基因组序列的长度例如为1.0kb以内、0.8kb以内、0.6kb以内、0.5kb以内、0.4kb以内、0.3kb以内、0.2kb以内或0.1kb以内。
连接多核苷酸中包含的阳性选择标记基因和阴性选择标记基因优选相互接近。通过使两者接近,可以防止在阳性选择标记基因与阴性选择标记基因之间发生重组(或者抑制到足够低的频率)。例如,在将从启动子至转录终止序列视为1个基因时,阳性选择标记基因与阴性选择标记基因之间例如为10kb以内,优选为8kb以内,更优选为7kb以内、5kb以内、4kb以内、3kb以内、2kb以内、1kb以内或0.5kb以内。
更优选从同一启动子转录阳性选择标记和阴性选择标记。因此,最优选阳性选择标记基因及阴性选择标记基因融合在一起,阳性选择标记和阴性选择标记表达为融合蛋白质。
如上所述,供体多核苷酸中包含的基因组片段在任意位置是可切割的。这里,可切割是指可以将该位置人工切割。切割优选在供体多核苷酸中的该位置唯一地发生,即,优选仅在供体多核苷酸中的该位置发生。
作为可切割的基因组片段,可以举出例如基因组片段中具有限制酶位点的情况。在该情况下,可以利用该限制酶切割该位置。限制酶位点优选仅包含在供体多核苷酸中的该位点。另外,如果在基因组片段中添加切割序列,则可以以该位点作为可切割位点。作为切割序列,没有特别限制,可以使用希望的限制酶的切割序列(例如noti位点)、大范围核酸酶的切割序列(i-scei位点、pi-scei位点等)、其它切割酶识别序列等。切割酶可以是天然的核酸酶,也可以是人工的核酸酶。另外,切割可以是单链切割,也可以是双链切割,优选为双链切割。在双链切割的情况下,切割位点可以形成平滑末端,也可以形成5’或3’突出末端。核酸酶可以适当添加核定位信号(nls)。实施例3所示的、添加于i-scei核酸酶的nh2末端的nls氨基酸序列为mdkaelipeppkkkrkvelgt(序列号42),但并不限定于该序列。
作为具体的一例,可以列举:来自于saccharomycescerevisiae(酿酒酵母)的归巢核酸内切酶i-scei(genbank:eu004203.1)或pi-scei(genbank:z74233.1)的切割序列,但并不限定于此。在将切割序列添加于供体多核苷酸中的基因组片段内的情况下,该序列优选不包含在与供体多核苷酸中包含的基因组片段对应的、成为供体多核苷酸的给予对象的细胞的对应基因组片段(即,与供体多核苷酸中包含的基因组片段对应的区域,所述基因组片段是在通过供体多核苷酸进行修饰之前的细胞所具有的基因组片段)的序列中。更优选可以合理地和/或统计地期望该切割序列也不包含在对象细胞的整个基因组中、或者仅以足够低频率(例如,以整个基因组计为10个部位以下、5个部位以下、3个部位以下、2个部位以下或1个部位以下)包含。
i-scei的切割序列是已知的,使用了5’-tagggataacagggtaat-3’18-bp(序列号1)的序列(colleaux,l.etal.recognitionandcleavagesiteoftheintron-encodedomegatransposase.proc.natl.acad.sci.usa85,6022-6026(1988))。需要说明的是,以该序列作查询(query)进行blastsearch,在数据库上的人基因组及转录产物中也没有发现与该序列匹配的序列。
供体多核苷酸的基因组片段中包含的1个或多个修饰部位(目标修饰部位)与可切割位点的位置关系没有特别限制,优选全部修饰部位集中在可切割位点的一侧。由此,在供体多核苷酸的切割位点附近发生与靶细胞的基因组的同源重组的情况下,多个修饰部位会重组在一起而不会被断开,因此可以通过一次操作获得导入了多个目标修饰部位的细胞。
如后所述,对于具有将供体多核苷酸导入细胞的基因组而得到的靶插入结构体的细胞而言,具有修饰部位的基因组和原本细胞所具有的基因组可以没有特定顺序地排列在连接多核苷酸的前后,但在该细胞进一步自然发生重组而去除连接多核苷酸时,位于其前后的基因组序列的一部分也被去除。本发明包括:在供体多核苷酸所包含的基因组片段中将可切割位点配置于修饰部位的上游侧(即,以作为修饰对象的基因的正义链而言为5’侧)或下游侧(即,以作为修饰对象的基因的正义链而言为3’侧)的任意方式。
供体多核苷酸中包含的基因组片段的长度没有特别限制,在导入了细胞的情况下,只要是用于发生与细胞基因组的同源重组的足够长度即可,可以使用希望长度的片段。供体多核苷酸中包含的基因组片段的长度例如为0.05kb以上、0.5kb以上、1kb以上、1.5kb以上、2kb以上、3kb以上、4kb以上或5kb以上,例如为10000kb以下、5000kb以下、500kb以下、300kb以下、200kb以下、100kb以下、80kb以下、50kb以下、30kb以下、20kb以下或10kb以下。需要说明的是,通过将供体多核苷酸中包含的基因组片段扩大至相当于一个基因座大小的数十kb或数百kb左右,也可以对大范围的多个突变部位一起进行修饰。本发明的供体多核苷酸可以包含这样长的基因组片段。
供体多核苷酸中包含的基因组片段的序列与靶细胞所对应的基因组序列具有很高的同一性,由此,可在与靶细胞的基因组之间诱导同源重组。例如,除修饰部位及切割位点以外的基因组片段的序列可以与靶细胞的基因组所对应的片段的序列具有通常90%以上、优选为95%以上、96%以上、97%以上、98%以上、99%以上或100%的同一性。
从供体多核苷酸中包含的基因组片段的可切割位点至与其最近的修饰部位的长度通常为10碱基以上、优选为20碱基以上、30碱基以上、40碱基以上、50碱基以上、80碱基以上、100碱基以上、200碱基以上、300碱基以上、400碱基以上、500碱基以上、800碱基以上或1kb以上。另外,通常为10000kb以内、5000kb以内、500kb以内、100kb以内,优选为80kb以内、70kb以内、60kb以内、50kb以内、40kb以内、30kb以内、20kb以内、10kb以内、8kb以内、7kb以内、6kb以内或5kb以内。另外,在供体多核苷酸中包含的基因组片段中,从与连接多核苷酸最近的修饰部位至与该连接多核苷酸的连接位点的长度也与上述相同。
更具体而言,从供体多核苷酸中包含的基因组片段的可切割位点至与其最近的修饰部位的长度例如优选为100碱基以上、150碱基以上、200碱基以上、250碱基以上、300碱基以上、316碱基以上、350碱基以上、400碱基以上、500碱基以上、600碱基以上、1000碱基以上、1200碱基以上、1500碱基以上或1960碱基以上。另外,在供体多核苷酸中包含的基因组片段中,从与连接多核苷酸最近的可切割位点至与该连接多核苷酸的连接位点的长度例如优选为200碱基以上、250碱基以上、300碱基以上、316碱基以上、350碱基以上、400碱基以上、500碱基以上、600碱基以上、1000碱基以上、1200碱基以上、1500碱基以上、1960碱基以上、2000碱基以上、2244碱基以上或2560碱基以上。
另外,本发明涉及使用本发明的供体多核苷酸修饰细胞的基因组序列的方法。该方法包括:(a)将本发明的供体多核苷酸导入细胞的工序、(b)通过阳性选择标记选择导入了该供体多核苷酸的细胞的工序、以及(c)通过阴性选择标记选择去除了该连接多核苷酸的细胞的工序。该方法例如可以在生物体外(例如体外(invitro)或离体(exvivo))实施。需要说明的是,在本发明中,体外实施也包括离体实施。
导入供体多核苷酸的细胞是基因组带有与供体多核苷酸中包含的基因组片段的序列具有高同一性的序列的细胞,通常是与供体多核苷酸中包含的基因组片段的来源生物相同物种的细胞。这样的细胞例如可以是希望的真核生物的细胞,例如为动物细胞或植物细胞,优选为动物细胞,更优选为哺乳动物细胞,例如为灵长类的细胞,具体可以举出小鼠、大鼠、猴及人的细胞。另外,细胞的种类没有特别限制,可以使用希望的组织的细胞,可以将供体多核苷酸导入已分化的细胞、未分化的细胞、前体细胞、祖细胞等进行基因组修饰。另外,也可以导入多能干细胞(例如诱导多能干细胞(ips细胞))等。
导入细胞的供体多核苷酸可以为环状,也可以为直链状。在导入直链状的供体多核苷酸的情况下,对供体多核苷酸中的基因组片段的可切割位点进行切割,形成直链状,导入细胞。该位点的切割方法没有特别限制,例如,只要是核酸酶的切割位点即可,可以用该核酸酶进行切割。优选在切割后为了去除核酸酶等而对供体多核苷酸进行纯化,或者将核酸酶灭活后导入细胞。
在导入环状的供体多核苷酸的情况下,在导入细胞时或在其后,供体多核苷酸中的基因组片段的可切割位点被切割。为了切割该位点,可以使切割该位点的核酸酶导入细胞或在细胞中表达。为了使核酸酶表达,例如,可以将编码该核酸酶的载体导入细胞。
在细胞中使核酸酶表达的情况下,其时机只要可使供体多核苷酸与核酸酶在细胞内接触而引起切割反应即可,没有限制,可以是将供体多核苷酸导入细胞之前、同时、或之后。优选在供体多核苷酸被导入细胞的前后48小时以内表达核酸酶,更优选在24小时以内表达。另外,优选在供体多核苷酸被导入细胞的前后48小时以内表达核酸酶,更优选在24小时以内表达。例如,通过将表达核酸酶的载体和供体多核苷酸同时导入细胞,可以高效地实现基因组编辑。在由载体表达核酸酶的情况下,考虑到表达充分升高为止的时滞,也可以在将供体多核苷酸导入细胞之前将载体导入细胞。
表达供体多核苷酸和/或核酸酶的载体向细胞的导入可以适当利用公知的方法来实施,没有特别限制。例如,可以利用脂质体转染、电穿孔、显微注射、基因枪法、以及病毒载体进行导入。
在为了核酸酶的表达而使用病毒载体的情况下,可以使用逆转录病毒载体、腺病毒载体、腺相关病毒载体、痘苗病毒载体等希望的病毒载体。在本发明中,作为特别优选使用的病毒载体,可以举出负链rna病毒载体,例如,可以优选使用副粘病毒载体。副粘病毒是指属于副粘病毒科(paramyxoviridae)的病毒或其衍生物。副粘病毒科包括副粘病毒亚科(paramyxovirinae)(包含呼吸道病毒属(也称为副粘病毒属)、腮腺炎病毒属及麻疹病毒属)及肺病毒亚科(pneumovirinae)(包含肺病毒属及变性肺病毒属)。作为副粘病毒科病毒中包含的病毒,具体可以列举:仙台病毒(sendaivirus)、新城疫病毒(newcastlediseasevirus)、腮腺炎病毒(mumpsvirus)、麻疹病毒(measlesvirus)、呼吸道合胞病毒(respiratorysyncytialvirus)、牛瘟病毒(rinderpestvirus)、犬瘟病毒(distempervirus)、猴副流感病毒(sv5)、人副流感病毒1、2、3型等。更具体而言,例如包括:仙台病毒(sendaivirus(sev))、人副流感病毒(humanparainfluenzavirus-1(hpiv-1))、人副流感病毒(humanparainfluenzavirus-3(hpiv-3))、海豹瘟病毒(phocinedistempervirus(pdv))、犬瘟热病毒(caninedistempervirus(cdv))、海豚麻疹病毒(dolphinmolbillivirus(dmv))、小型反刍动物瘟疫病毒(peste-des-petits-ruminantsvirus(pdpr))、麻疹病毒(measlesvirus(mev))、牛瘟病毒(rinderpestvirus(rpv))、亨德拉病毒(hendravirus(hendra))、立白病毒(nipahvirus(nipah))、人副流感病毒(humanparainfluenzavirus-2(hpiv-2))、猴副流感病毒(simianparainfluenzavirus5(sv5))、人副流感病毒(humanparainfluenzavirus-4a(hpiv-4a))、人副流感病毒(humanparainfluenzavirus-4b(hpiv-4b))、腮腺炎病毒(mumpsvirus(mumps))、以及新城疫病毒(newcastlediseasevirus(ndv))等。作为弹状病毒,包括弹状病毒科(rhabdoviridae)的水疱性口炎病毒(vesicularstomatitisvirus)、狂犬病毒(rabiesvirus)等。
另外,负链rna病毒可以来自天然株、野生株、突变株、实验室传代株、以及人工构建的毒株等。例如,在仙台病毒的情况下,可以举出z株,但并不限定于此(medicaljournalofosakauniversityvol.6,no.1,march1955p1-15)。例如,可以是野生型病毒所具有的任意基因中具有突变、缺失的病毒。例如,可以优选使用传播能力缺失型病毒,所述传播能力缺失型病毒在编码病毒的包膜蛋白或外壳蛋白的至少1个基因中具有缺失或抑制其表达的终止密码子突变等突变。这样的不表达包膜蛋白的病毒例如是虽然可以在感染细胞中复制基因组但无法形成感染性病毒粒子的病毒。这样的传播能力缺失型的病毒特别适合作为高安全性的载体。例如,可以使用基因组中不编码f或hn中的任意包膜蛋白(刺突蛋白)的基因、或者f及hn的基因的病毒wo00/70055及wo00/70070;li,h.-o.etal.,j.virol.74(14)6564-6569(2000))。只要基因组rna中至少编码了基因组复制所需要的蛋白质(例如n、p及l蛋白),病毒就可以在感染细胞中扩增基因组。为了制造包膜蛋白缺失型且具有感染性的病毒粒子,例如,在病毒产生细胞中,外源性地供给缺失了的基因产物或能够对其互补的蛋白质(wo00/70055及wo00/70070;li,h.-o.etal.,j.virol.74(14)6564-6569(2000))。另一方面,通过完全不互补缺失的病毒蛋白质,可以回收非感染性病毒粒子(wo00/70070)。
另外,作为病毒载体,也优选使用装载突变型的病毒蛋白质基因的病毒载体。例如,在包膜蛋白、外壳蛋白中,已知有包括减毒化突变、温度敏感性突变的多种突变。在本发明中可以优选使用具有这些突变蛋白质基因的病毒。在本发明中,可以优选使用减弱了细胞毒性的载体。例如,在病毒的结构蛋白(np,m)、rna合成酶(p,l)中,已知有包含减毒化突变、温度敏感性突变的多种突变。在本发明中,可以根据目的优选使用具有这些突变蛋白质基因的副粘病毒载体等。
具体而言,例如,作为仙台病毒的m基因的优选突变,可以举出从m蛋白中的69位(g69)、116位(t116)及183位(a183)中任意选择的位点的氨基酸取代(inoue,m.etal.,j.virol.2003,77:3238-3246)。在本发明中,优选使用具有编码突变m蛋白的基因组的病毒,所述突变m蛋白是在仙台病毒的m蛋白中上述3个位点中的任一个、优选为任意2个位点的组合、进一步优选为全部3个位点的氨基酸被取代为其它氨基酸而成的。
氨基酸突变优选为侧链取代成化学性质不同的其它氨基酸,例如,取代为blosum62矩阵(henikoff,s.andhenikoff,j.g.(1992)proc.natl.acad.sci.usa89:10915-10919)的值为3以下、优选为2以下、更优选为1以下、更优选为0的氨基酸。具体而言,可以将仙台病毒m蛋白的g69、t116及a183分别取代为glu(e)、ala(a)及ser(s)。另外,也可以利用与麻疹病毒温度敏感性株p253-505(morikawa,y.etal.,kitasatoarch.exp.med.1991:64;15-30)的m蛋白的突变同源的突变。突变的导入可以使用例如寡核苷酸等按照公知的突变导入方法来实施。
另外,作为hn基因的优选突变,例如可以举出从仙台病毒的hn蛋白的262位(a262)、264位(g264)及461位(k461)中任意选择的位点的氨基酸取代(inoue,m.etal.,j.virol.2003,77:3238-3246)。在本发明中,优选使用具有编码突变hn蛋白的基因组的病毒,所述突变hn蛋白是上述的3个位点中的任1个、优选为任意2个位点的组合、进一步优选为全部3个位点的氨基酸被取代为其它氨基酸而成的。与上述相同,氨基酸的取代优选为侧链取代成化学性质不同的其它氨基酸。作为一个优选的例子,将仙台病毒hn蛋白的a262、g264及k461分别取代为thr(t)、arg(r)及gly(g)。另外,例如,以腮腺炎病毒的温度敏感性疫苗株urabeam9为参考,可以对hn蛋白的第464及468位的氨基酸进行突变导入(wright,k.e.etal.,virusres.2000:67;49-57)。
另外,仙台病毒可以在p基因和/或l基因具有突变。作为这样的突变,具体可以列举:sevp蛋白的第86位的glu(e86)的突变、sevp蛋白的第511位leu(l511)取代为其它氨基酸。与上述相同,氨基酸的取代优选为侧链取代成化学性质不同的其它氨基酸。具体可以示例出:第86位的氨基酸取代成lys、第511位的氨基酸取代为phe等。另外,在l蛋白中,可以举出sevl蛋白的第1197位的asn(n1197)和/或第1795位的lys(k1795)取代为其它氨基酸,与上述相同,氨基酸的取代优选为侧链取代成化学性质不同的其它氨基酸。具体可以示例出:第1197位的氨基酸取代成ser、第1795位的氨基酸取代成glu等。p基因及l基因的突变可以显著提高持续感染性、抑制2次粒子释放、或抑制细胞毒性的效果。另外,通过将包膜蛋白基因的突变和/或缺失组合,可以明显提升这些效果。另外,l基因可以举出sevl蛋白的第1214位的tyr(y1214)和/或第1602位的met(m1602)取代为其它氨基酸,与上述相同,氨基酸的取代优选为侧链取代成化学性质不同的其它氨基酸。具体可以示例出:第1214位的氨基酸取代成phe、第1602位的氨基酸取代成leu等。以上示例出的突变可以任意组合。
例如,在本发明中,以下仙台病毒载体是合适的:sevm蛋白的至少69位的g、116位的t及183位的a、sevhn蛋白的至少262位的a、264位的g及461位的k、sevp蛋白的至少511位的l、sevl蛋白的至少1197位的n及1795位的k分别取代为其它氨基酸、且缺损或缺失f基因的仙台病毒载体;以及细胞毒性与它们相同或更低、和/或37℃下对ntvlp形成的抑制与它们相同或更高的缺损或缺失f基因的仙台病毒载体。
更具体而言,在本发明中可以优选使用缺失f基因、且于基因组中在m蛋白包含g69e、t116a及a183s的突变、在hn蛋白包含a262t、g264r及k461g的突变、在p蛋白包含l511f的突变、以及在l蛋白包含n1197s及k1795e的突变的仙台病毒载体。在本发明中,将f基因的缺失和这些突变的组合称为“tsδf”。
另外,例如在仙台病毒(sev)的情况下,作为l蛋白的突变,可以举出从sevl蛋白的942位(y942)、1361位(l1361)及1558位(l1558)中任意选择的位点的氨基酸取代为其它氨基酸。与上述相同,氨基酸的取代优选为侧链取代成化学性质不同的其它氨基酸。具体可以示例出第942位的氨基酸取代成his、第1361位的氨基酸取代成cys、第1558位的氨基酸取代成ile等。特别可以优选使用至少942位或1558位被取代的l蛋白。例如,除1558位以外,1361位也被取代成其它氨基酸的突变l蛋白也是优选的。另外,除942位以外,1558位和/或1361位也被取代成其它氨基酸的突变l蛋白也是优选的。通过这些突变,可以提高l蛋白的温度敏感性。
另外,作为p蛋白的突变,可以举出从sevp蛋白的433位(d433)、434位(r434)及437位(k437)中任意选择的位点的氨基酸取代为其它氨基酸。与上述相同,氨基酸的取代优选为侧链取代成化学性质不同的其它氨基酸。具体可以示例出第433位的氨基酸取代成ala(a)、第434位的氨基酸取代成ala(a)、第437位的氨基酸取代成ala(a)。特别可以优选使用全部这些3个位点被取代的p蛋白。通过这些突变,可以提高p蛋白的温度敏感性。
本发明中也可以优选使用以下仙台病毒载体:编码sevp蛋白的至少433位的d、434位的r及437位的k这3个部位取代成其它氨基酸的突变p蛋白、以及sevl蛋白的至少1558位的l被取代的突变l蛋白(优选为至少1361位的l也被取代为其它氨基酸的突变l蛋白)、且缺损或缺失f基因的仙台病毒载体;以及细胞毒性与它们相同或更低、和/或温度敏感性与它们相同或更高的缺损或缺失f基因的仙台病毒载体。各病毒蛋白除本说明书中示例出的突变以外,还可以在其它氨基酸(例如10个以内、5个以内、4个以内、3个以内、2个以内、或1个氨基酸)具有突变。由于具有上述所示的突变的载体显示出很高的温度敏感性,因此可以通过在通常温度(例如约37℃、具体为36.5~37.5℃、优选为36.6~37.4℃、更优选为36.7℃~37.3℃)下培养细胞来简便地去除载体。在载体的去除中,可以在稍高温(例如37.5~39℃、优选为38~39℃、或38.5~39℃)下进行培养。
示例出具体的载体,可以列举例如:缺失f基因、且于基因组中在m蛋白包含g69e、t116a及a183s的突变、在hn蛋白包含a262t、g264r及k461g的突变、在p蛋白包含l511f的突变、以及在l蛋白包含n1197s及k1795e的突变的仙台病毒载体。
另外,优选可以列举例如:缺失f基因、且于基因组中在m蛋白包含g69e、t116a及a183s的突变、在hn蛋白包含a262t、g264r及k461g的突变、在p蛋白包含l511f的突变、以及在l蛋白包含n1197s及k1795e的突变、并且在基因组中进一步包含以下(i)和/或(ii)的突变的仙台病毒载体。
(i)p蛋白的d433a、r434a及k437a的突变
(ii)l蛋白的y942h、l1361c和/或l1558i的突变
作为更具体的示例,可以列举:缺失f基因、且于基因组中在m蛋白包含g69e、t116a及a183s的突变、在hn蛋白包含a262t、g264r及k461g的突变、在p蛋白包含l511f的突变、以及在l蛋白包含n1197s及k1795e的突变、并且在基因组中进一步包含以下(i)~(iv)中任一种突变的仙台病毒载体。
(i)p蛋白的d433a、r434a及k437a的突变、以及l蛋白的l1361c及l1558i的突变(ts15)
(ii)p蛋白的d433a、r434a及k437a的突变(ts12)
(iii)l蛋白的y942h、l1361c及l1558i的突变(ts7)
(iv)p蛋白的d433a、r434a及k437a的突变、以及l蛋白的l1558i的突变(ts13)
(v)p蛋白的d433a、r434a及k437a的突变、以及l蛋白的l1361c的突变(ts14)
在使核酸酶基因装载于载体时,核酸酶基因可以在紧邻任意病毒基因(np、p、m、f、hn或l)之前(基因组的3’侧)或之后(基因组的5’侧)插入。例如,可以在紧邻仙台病毒的p基因之后、即紧邻p基因的下游(紧邻负链rna基因组的5’侧)导入,但并不限定于此。
例如,负链rna病毒的制造可以利用以下的公知的方法来实施(wo97/16539;wo97/16538;wo00/70055;wo00/70070;wo01/18223;wo03/025570;wo2005/071092;wo2006/137517;wo2007/083644;wo2008/007581;hasan,m.k.etal.,j.gen.virol.78:2813-2820,1997、kato,a.etal.,1997,emboj.16:578-587及yu,d.etal.,1997,genescells2:457-466;durbin,a.p.etal.,1997,virology235:323-332;whelan,s.p.etal.,1995,proc.natl.acad.sci.usa92:8388-8392;schnell.m.j.etal.,1994,emboj.13:4195-4203;radecke,f.etal.,1995,emboj.14:5773-5784;lawson,n.d.etal.,proc.natl.acad.sci.usa92:4477-4481;garcin,d.etal.,1995,emboj.14:6087-6094;kato,a.etal.,1996,genescells1:569-579;baron,m.d.andbarrett,t.,1997,j.virol.71:1265-1271;bridgen,a.andelliott,r.m.,1996,proc.natl.acad.sci.usa93:15400-15404;tokusumi,t.etal.virusres.2002:86;33-38、li,h.-o.etal.,j.virol.2000:74;6564-6569)。另外,对于病毒的增殖方法及重组病毒的制造方法,可以参照病毒学实验学各论、修正二版(国立预防卫生研究所学友会编、丸善、1982)。
在本发明的方法中,在将供体多核苷酸导入细胞的工序(工序(a))之后,通过阳性选择标记选择保持该连接多核苷酸的细胞(即,保持供体多核苷酸的细胞)(工序(b))。该工序可以根据标记的种类来适当实施。例如,在使用了药物抗性标记的情况下,将细胞与该药物一起培养,选择表达标记的细胞。细胞的选择可以是将表达阳性选择标记的细胞群从不表达的细胞群中完全分离,另外,也可以是使表达阳性选择标记的细胞的比例增加。通过该选择,全部细胞中的阳性选择标记阳性细胞(或导入了供体多核苷酸的细胞)的比例显著升高。例如,通过该选择,全部细胞中的阳性选择标记阳性细胞(或导入了供体多核苷酸的细胞)的比例升高10倍以上、50倍以上、100倍以上、500倍以上、1000倍以上、5000倍以上、10000倍以上、50000倍以上或10万倍以上。另外,通过该选择,全部细胞中的阳性选择标记阳性细胞(或导入了供体多核苷酸的细胞)的比例优选为0.0000001以上、0.000001以上、0.00001以上、0.0001以上、0.001以上、0.01以上、0.02以上、0.05以上、0.1以上、0.2以上、0.3以上、0.4以上、0.5以上、0.6以上、0.7以上、0.8以上、0.9以上、0.95以上、0.98以上或0.99以上。
在本发明的供体多核苷酸通过同源重组引入细胞的基因组时,该区域具有以下结构:(a)细胞原本具有的基因组片段-(b)供体多核苷酸中包含的基因组片段-(c)供体多核苷酸中包含的连接多核苷酸-(d)供体多核苷酸中包含的基因组片段-(e)细胞原本具有的基因组片段。但是,在供体多核苷酸中包含的基因组片段除目标修饰部位以外与细胞的基因组相同的情况下,看起来具有如下结构:经由供体多核苷酸中包含的连接多核苷酸,基本上相同的基因组序列排列在连接多核苷酸的前后。在本发明中,将这样的结构体称为供体多核苷酸的靶插入结构体。在经由连接多核苷酸排列在前后的基因组片段的哪个中包含目标修饰(即供体多核苷酸中包含的修饰序列)可以根据同源重组发生的位置而改变,但均形成包含目标修饰的基因组片段和细胞原本具有的基因组片段经由连接多核苷酸串联排列的结构。另外,连接多核苷酸包含阳性选择标记基因及阴性选择标记基因。即,通过本发明的方法获得的、具有在基因组中引入了本发明的供体多核苷酸的结构的细胞是如下细胞:具有供体多核苷酸中包含的修饰基因组片段和来自于与其相对应的细胞的基因组的片段经由供体多核苷酸中包含的连接多核苷酸无特定顺序地串联而成的结构、且在该连接多核苷酸中包含阳性选择标记基因及阴性选择标记基因。
在本发明的方法中,接下来,通过阴性选择标记选择去除了该连接多核苷酸的细胞。如上所述,在本发明的供体多核苷酸通过同源重组导入细胞d基因组时,成为包含目标修饰的基因组片段和细胞原本具有的基因组片段经由连接多核苷酸串联排列的结构。由于这一对序列具有高同源性,因此能以高效率诱导同源重组,其结果是将包含连接多核苷酸的片段从基因组中去除。不需要进行特别的操作来引起该反应。通过对细胞进行培养,生成去除了连接多核苷酸的细胞。然后,通过利用连接多核苷酸中的阴性选择标记基因,可以主动选择从基因组中去除了连接多核苷酸的细胞。
该工序也可以根据阴性选择标记的种类而适当实施。例如,在使用了胸苷激酶(tk)基因的情况下,将细胞和更昔洛韦一起培养,选择不表达标记的细胞(或去除了连接多核苷酸的细胞)。细胞的选择可以是将不表达阴性选择标记的细胞(或去除了连接多核苷酸的细胞)的群从表达阴性选择标记的细胞(或去除了连接多核苷酸的细胞)的群中完全分离,另外,也可以是使不表达阴性选择标记的细胞(或去除了连接多核苷酸的细胞)的比例增加。通过该选择,全部细胞中的不表达阴性选择标记的细胞(或去除了连接多核苷酸的细胞)的比例显著升高。例如,通过该选择,全部细胞中的不表达阴性选择标记的细胞(或去除了连接多核苷酸的细胞)的比例升高10倍以上、50倍以上、100倍以上、500倍以上、1000倍以上、5000倍以上、10000倍以上、50000倍以上、或10万倍以上。另外,通过该选择,全部细胞中的不表达阴性选择标记的细胞(或去除了连接多核苷酸的细胞)的比例优选为0.00001以上、0.0001以上、0.001以上、0.01以上、0.02以上、0.05以上、0.1以上、0.2以上、0.3以上、0.4以上、0.5以上、0.6以上、0.7以上、0.8以上、0.9以上、0.95以上、0.98以上或0.99以上。
对于通过利用阴性选择标记进行选择而得到的细胞而言,通过同源重组在连接多核苷酸的前后串联排列的基因组片段恢复为单拷贝。虽然希望得到的细胞的连接多核苷酸部分被全部去除,但重复的基因组序列区域的哪个部分被去除会随重组发生的位置而改变。因此,得到的细胞中混合存在导入了目标基因组修饰的细胞和未导入的细胞(即恢复为原始状态的细胞)。但是,由于这些细胞基本上以同等的概率出现,因此可以容易地获得保持目标修饰的细胞。是否具有目标修饰例如可以通过直接或间接地检测对目标修饰部位特异性的序列来进行,例如,可以通过pcr扩增靶位点并确认碱基序列,或者进行利用了突变位点特异性引物等的pcr,通过确认有无pcr产物、扩增片段的长度等来识别。
以下,对本发明的基因修饰更详细地进行说明。
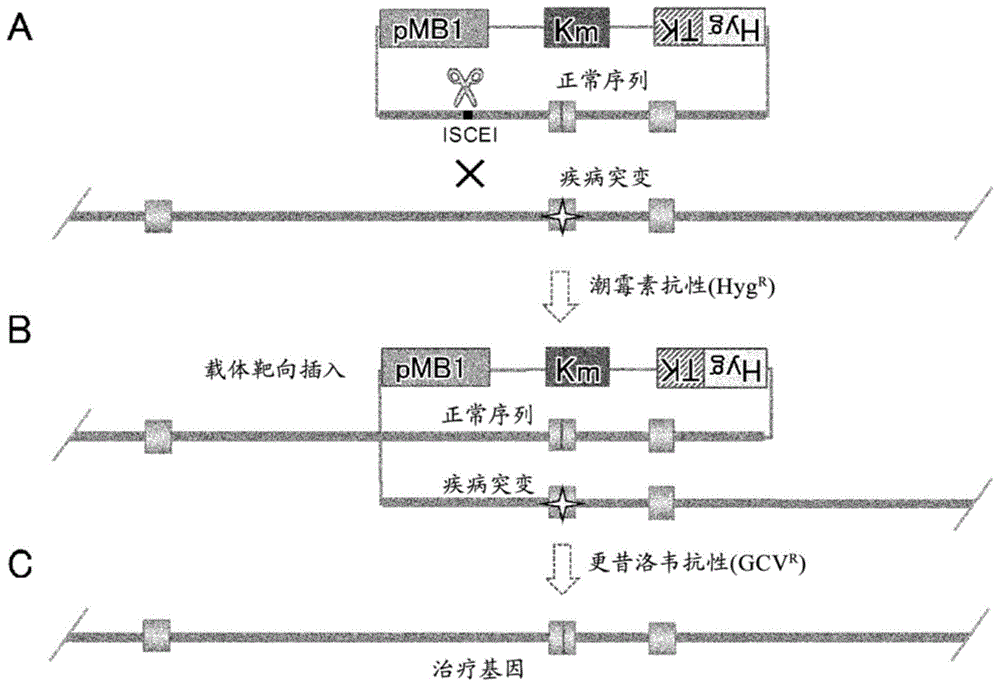
图1示出了利用本发明的一个实施方式进行基因修饰的方案。需要说明的是,这里,将供体多核苷酸也称为载体或供体质粒等。在第1阶段中(图1a-b),在靶基因内构建载体靶向插入体结构,所述载体靶向插入体结构是通过修饰后的碱基序列和修饰前的碱基序列夹住供体质粒的骨架区域而使修饰后碱基序列与修饰前碱基序列(无特定顺序)串联排列而成的,在第2阶段中(图1b-c),通过选择从该载体靶向插入体结构自发地取代为仅由修饰后碱基序列形成的单独结构的克隆,将修饰前碱基序列转变为修饰后碱基序列,从而进行基因的修饰。
在a中,用细线表示供体质粒的载体骨架,用粗线表示与供体质粒的修饰后序列位于中央附近的修饰基因同源的序列,在该质粒下方示出了细胞的想要修饰的基因座,示出了向修饰基因座的细胞导入供体质粒时,修饰基因座和供体质粒的同源区域彼此对齐。细胞所具有的基因组序列中包含了疾病突变,供体质粒所具有的基因组片段具有正常序列。
在b中,在同源区域对齐后,从由i-scei等形成的切断端开始同源重组反应,其结果是形成载体靶向插入体。该结构表明,包含修饰后序列(正常序列)的片段、包含修饰前序列(疾病突变)的片段夹住载体骨架(连接多核苷酸)而串联排列。将该载体靶向插入体细胞克隆分离为例如潮霉素抗性克隆。
在c中示出了通过从该载体靶向插入体结构自发地取代为仅由修饰后碱基序列形成的单独结构,从而使修饰前碱基序列转变为修饰后碱基序列。将该取代细胞克隆分离为例如更昔洛韦抗性克隆。由此,具有作为上述第1阶段产物的载体靶向插入体结构(即,包含修饰后序列(正常序列)的片段和包含修饰前序列(疾病突变)的片段夹住载体骨架(连接多核苷酸)串联排列的结构)的细胞是通过修饰前碱基序列被自发地取代为修饰后碱基序列而可以转变为仅由修饰后碱基序列序列形成的单独结构的、具有载体靶向插入体结构的细胞株。
本申请的实施例中使用的供体多核苷酸的载体骨架含有大肠杆菌dna复制的起始位点(pmb1)、卡那霉素抗性基因(大肠杆菌选择标记;km)、人转录起始位点控制下的由潮霉素抗性基因(动物细胞选择标记)和hsv-tk(动物细胞排除标记)形成的融合基因(hygtk)。与修饰基因同源的序列由跨越人hprt基因的内含子1、具有修饰后序列(正常序列)的外显子2、内含子2、外显子3、内含子3的5488bp的片段形成,修饰基因座在外显子2内部具有作为修饰对象的修饰前序列(疾病突变;以十字形的星表示)。
图2中示出了利用本发明的基因组编辑技术进行基因修饰的验证实验的步骤。a-c示出了与图1相同的基因修饰方案,由于供体质粒具有将外显子2上的sexa1位点(accaggt)附近进行了同义转换的分子标记,因此可以通过从sexa1位点至同义转换序列的改变来确认基因座靶区域的序列修饰。d-f示出了对a-c的反应的操作步骤。将通过位点特异性切割酶i-scei处理的直链上供体质粒(同义转换序列)导入靶基因座(sexa1位点)的细胞,通过pcr筛选从潮霉素抗性细胞集落获得载体靶向插入体细胞,在将该克隆培养/接种后,得到更昔洛韦抗性细胞集落,获得基因修饰体。
这样,为了得到具有作为本发明方法的第1阶段产物的载体靶向插入体结构的细胞,对于修饰前碱基序列位点可以使用在修饰后碱基序列内的5’侧具有单独dna切割位点、且在质粒的骨架区域具有阳性选择标记基因和阴性选择标记(排除标记)基因的供体质粒dna(参照图1a)、或者在修饰后碱基序列内的3’侧具有单独dna切割位点、且在质粒的骨架区域具有阳性选择标记基因和阴性选择标记基因的供体质粒dna(参照图2a)。这里,5’侧及3’侧分别是指以作为修饰对象的基因的正义链而言的5’侧及3’侧。
图3示出了载体靶向插入体的pcr筛选的方法。在a中示出了载体靶向插入的区域的结构。来自供体质粒的修饰后序列(同义转换序列)位于下游,来自靶基因座的修饰前序列(sexa1序列)位于上游。如b左图所示,如果根据利用载体靶细胞的基因组dna、上游5488bp片段的5’外侧的引物gt68和位于载体骨架的hygtk启动子区域的引物gt124的pcr检测到了相当于7577bp的片段,则可以认为其是载体靶向插入体的上游区域(5’区域)的结构。同样地如b右图所示,如果根据利用位于载体骨架的pmb1区域的引物gt112和下游5488bp片段的3’外侧的引物gt68的pcr检测到了相当于7683bp的片段,则可以认为其是载体靶向插入体的下游区域(3’区域)的结构。另外,为了确认上游区域的sexa1序列和下游区域的同义转换序列共存,根据利用对分子标记序列所在的外显子2的区域进行扩增的引物gt19/gt22的pcr,得到相当于525bp的片段并进行序列分析,根据得到的波形图确认2种标记序列共存。
图4示出了基于更昔洛韦选择从载体靶向插入体分离基因修饰体的方法。a中示出,在发生从图3a的载体靶向插入体取代为上游(重复的前半)或下游(重复的后半)序列的反应时,与此相伴,释放出各具有下游序列或上游序列的载体质粒,但通过更昔洛韦选择,具有释放出的质粒的细胞被排除,得到在基因座发生了取代反应的基因修饰体的候选克隆。如b右图所示,为了确认更昔洛韦抗性克隆的基因座为取代体结构,根据利用同源基因座区域5488bp片段的5’外侧引物gt68和3’外侧引物gt69进行的pcr分析,如果检测到相当于9367bp的片段,则可以认为其是基因修饰体的结构。如b中图所示,为了确认更昔洛韦抗性克隆的细胞内不存在释放出的质粒,根据利用对质粒上hygtk基因的hyg区域进行扩增的引物gt38/gt39进行的pcr分析,如果没有产生相当于998bp的片段,则可以判定该细胞克隆中不包含释放出的质粒。最后,如b右图所示,为了从更昔洛韦抗性克隆中分离基因修饰体,根据利用对分子标记序列所在的外显子2的区域进行扩增的引物gt19/gt22的pcr,得到相当于525bp的片段并确定序列,该图显示可以得到在载体靶向插入区域上的重复的修饰前序列(sexa1序列)和修饰后序列(同义转换序列)中取代为修饰后序列的细胞克隆。另外,不仅是供体质粒的切割为3’侧的形式,而且5’侧切割也是本发明的具体实施方式。需要说明的是,重复区域中的修饰部位前后的长度差异没有问题。即,位于修饰部位之前(基因的编码链的5’侧)的重复区域可以比位于后面(3’侧)的重复区域长,位于前面的重复区域也可以比位于后面的重复区域短,两者也可以为相同长度。
图5示出了以往基因修饰方法的问题和本发明的方法的优越性。a中示出了发明要解决的课题。c的“课题项目”列中列举了上述的on/off-target不正确再结合的课题、位点特异性重组位点残留的课题、失去靶切割序列的问题这样的待解决的课题。b中示出了解决该课题的方法。如c所示,通过使用本发明的方法,可以避免on/off-target不正确再结合、位点特异性重组位点(loxp等)的残留、以及失去靶切割序列等问题。
本发明可用于在细胞的基因组中导入希望的修饰,例如,在遗传性疾病的致病基因中,可以用于将致病序列转变为正常序列。例如,作为本发明的疾病突变修复方法的一例,可以举出如下基因修复方法:在第1阶段,不发生治疗对象的遗传性疾病的正常碱基序列和作为治疗对象的致病碱基序列夹住供体多核苷酸中的连接多核苷酸(例如质粒的骨架区域),在靶致病基因内构建正常碱基序列和致病碱基序列(无特定顺序)串联排列的载体靶向插入体结构,在第2阶段,通过从该载体靶向插入体结构自发地取代为仅由正常碱基序列形成的单独结构,将靶致病碱基序列转变为正常碱基序列。
作为用于疾病突变修复的供体多核苷酸的一例,为了得到具有上述疾病突变修复方法中第1阶段产物的载体靶向插入体结构的细胞,可以列举:在位于致病碱基序列位点的5’侧(基因的正义链的5’侧)的正常碱基序列内具有单独dna切割位点、且在供体多核苷酸的连接多核苷酸(例如质粒的骨架区域)具有选择标记(阳性选择标记)基因和排除标记(阴性选择标记)基因的供体多核苷酸(参照图1a);或者在位于致病碱基序列位点的3’侧(基因的正义链的3’侧)的正常基序列内具有单独dna切割位点、且在连接多核苷酸中(例如质粒的骨架区域)具有选择标记基因和排除标记基因的供体多核苷酸(参照图2a)。
通过将供体多核苷酸导入具有疾病突变的细胞,可以得到疾病突变修复载体靶向插入体细胞。该细胞是具有载体靶向插入体结构的细胞株,其通过将致病碱基序列自发地取代成正常碱基序列,从而可以由作为上述的疾病突变修复方法中第1阶段产物的载体靶向插入体结构转变为仅由正常碱基序列形成的单独结构。即,该细胞株是染色体上具有载体靶向插入体结构的细胞,所述载体靶向插入体结构是不发生治疗对象的遗传性疾病的正常碱基序列和作为治疗对象的致病碱基序列夹住供体多核苷酸中的连接多核苷酸(例如质粒的骨架区域)、且无特定顺序(即任意顺序)地串联排列而成的。在该细胞株中,该载体靶向插入体结构可自发且随机地被取代为仅由正常碱基序列形成的单独结构。
另外,本发明提供使用本发明的供体多核苷酸的单基因座取代方法。即使遗传性疾病为单基因性疾病,也存在序列、结构、尺寸方面的突变多样性,为了用一种供体多核苷酸分别对这样的多样疾病突变进行基因座取代,通过将本发明的供体多核苷酸扩大至相当于单基因座尺寸的数十kb/数百kb左右,提供如下的基因修复方法:在第1阶段,在靶致病基因座构建载体靶向插入体结构,所述载体靶向插入体结构是不发生治疗对象的遗传性疾病的正常基因座和作为治疗对象的一个致病突变基因座夹住供体多核苷酸中的连接多核苷酸(例如质粒骨架区域)、且正常基因座和致病突变基因座(无特定顺序)串联排列而成的,在第2阶段,通过从该载体靶向插入体结构自发地取代为仅由正常基因座形成的单独结构,将靶致病突变基因座转变为正常基因座。
另外,本发明还涉及在上述的单基因座取代方法中使用的单基因座取代供体多核苷酸。具体而言,为了得到具有作为上述单基因座取代方法中第1阶段产物的载体靶向插入体结构的细胞,该供体多核苷酸是相对于致病突变位点在正常基因座的5’侧具有单独dna切割位点、且连接多核苷酸中(例如质粒的骨架区域)具有选择标记基因和排除标记基因的供体多核苷酸,而且所述供体多核苷酸是与用于上述的疾病突变修复的供体多核苷酸相比为相同类型且更大型的供体多核苷酸(参照图1a)、或者在正常基因座的3’侧具有单独dna切割、且在连接多核苷酸中(例如质粒的骨架区域)具有选择标记基因和排除标记基因的供体多核苷酸(参照图2a)。
通过将该供体多核苷酸导入进行单基因座取代的对象细胞,可以得到单基因座取代载体靶向插入体细胞。该细胞是通过致病突变基因座被自发地转变为正常基因座而可以由作为上述单基因座取代方法中第1阶段产物的载体靶向插入体结构转变为仅由正常基因座形成的单独结构、且与作为上述疾病突变修复方法的第1阶段产物的载体靶向插入体结构相比具有同类型且更大型的载体靶向插入体结构的细胞株。
另外,本发明对于缩短将供体多核苷酸导入对象细胞并通过供体多核苷酸的靶插入体细胞获得具有单独结构的细胞的工序也是有用的。具体而言,例如,在供体多核苷酸具有质粒载体骨架的情况下,在对该质粒dna从基因组解离而分离、或插入了基因组的细胞进行选择培养中,从具有该细胞群中包含的靶区域中的载体靶向插入体结构的细胞自发地产生被取代为由上游或下游形成的单独结构的细胞时,对释放出的供体质粒dna的基因组随机插入细胞(例如,在非靶基因位点的部位插入了供体质粒dna序列的细胞)进行阴性选择培养,选择具有单独结构的细胞,筛选由修饰后序列形成的克隆。
本发明的供体多核苷酸可以适当地和药学上允许的载体或介质一起制成组合物。包含本发明的供体多核苷酸的组合物可以作为医药组合物、例如遗传疾病的基因组修复用的医药组合物来使用。载体及介质没有特别限制,可以列举例如:水(例如无菌水)、生理盐水(例如磷酸缓冲生理盐水)、乙醇、甘油、乳糖、蔗糖、磷酸钙、明胶、葡聚糖、琼脂、果胶、花生油、芝麻油等,还可以列举:缓冲液、双重稀释剂(doublediluents)、赋形剂、佐剂等。
给药途径可以适当确定,没有特别限定。本发明的供体多核苷酸或组合物可以在体内或离体使用,另外,例如可以通过肌肉内注射、静脉内、经皮、鼻腔内、腹腔内、口服、粘膜、或其它递送途径给药。给药次数及用量没有限制,可以是单次用途,也可以是多次给药。本领域技术人员可以根据组合物的种类、靶细胞、给药对象、组织、疾病、治疗对象的症状、状态、给药途径、给药方法等而适当选择。给药对象例如为哺乳动物(包括人及非人哺乳类),具体包括:人、猴等非人灵长类、小鼠、大鼠等啮齿类、兔、山羊、绵羊、猪、牛、犬、猫等其它全部哺乳动物。
另外,本发明涉及包含本发明的供体多核苷酸的试剂盒。该试剂盒可以包含本发明的供体多核苷酸、和对该供体多核苷酸中的基因组片段的可切割部位进行切割的切割酶或编码其的载体。另外,该试剂盒可以适当附带操作说明书。本发明的试剂盒对于使用了本发明的供体多核苷酸的基因组修饰是有用的。
作为载体,可以使用希望的载体,可以列举例如:质粒载体、病毒载体等,作为病毒载体,可以列举例如负链rna病毒载体,特别是副粘病毒载体,其中可举出仙台病毒载体。
实施例
以下,通过实施例对本发明更详细地进行说明,但本发明并不限于这些实施例。另外,本说明书中引用的文献及其它参照均作为本说明书的一部分而引入本说明书。
[实施例1]供体多核苷酸的制作
pmb1kmhygtk-hprtex2syn(1200)iscei(参照图2a)的构建
将实施例中使用的供体质粒载体的制作方法示于以下。
在本发明中,“pmb1kmhygtk”表示供体质粒载体的骨架。载体骨架中的“pmb1”表示开始大肠杆菌dna复制所必要的区域,“km”表示作为大肠杆菌选择标记的卡那霉素抗性基因,“hygtk”表示作为人转录起始区域之一的hef1-htlv启动子控制下的、由潮霉素抗性基因(动物细胞选择标记)和hsv-tk(动物细胞排除标记)形成的融合基因。
将与包含插入载体骨架的修饰后序列的修饰对象基因座同源的序列接着载体骨架之后的连字符表示为“-hprtex2syn(1200)iscei”。“hprtex2”表示跨越人hprt基因的内含子1、外显子2、内含子2、外显子3、内含子3的片段,另外,“ex2syn”表示修饰后序列syn包含于ex2内,“(1200)iscei”表示在从修饰后序列syn向3’侧1200bp的位置存在i-scei切割酶识别序列。在其它示例中,将与包含插入载体骨架的修饰后序列的修饰对象基因座同源的序列接着载体骨架之后的连字符表示为“-hprtiscei(1200)ex2syn”。“ex2syn”表示修饰后序列syn包含于ex2内,“iscei(1200)”表示在从修饰后序列syn向5’侧1200bp的位置存在i-scei切割酶识别序列。
但是,这些是示例,本发明并不限定于此。
pmb1kmhygtk供体质粒(参照图2a)的构建
如下所述进行了pselect-km-hsv1tk亚质粒的构建。以pcr-bluntii-topoplasmiddna(invitrogen)作为模板,使用5’-cttaattaacctgcagccggaattgccagctg-3’(gt82)(序列号2)及5’-atgtggtatggaattcggtggccctcctcacgtgc-3’(gt83)(序列号3),进行利用kod-plus-dnapolymerase(toyobo)的pcr反应(94℃-2分钟→将94℃-15秒钟、55℃-30秒钟、68℃-1分30秒钟进行40个循环→68℃7分钟),得到了约1000碱基的pcr产物。利用in-fusionkit(toyobo)使上述1000-basepcr产物与用ecori限制酶和psti限制酶消化的pselect-zeo-hsv1tk连接,得到了pselect-km-hsv1tk(1)。
如下所述进行了pselect-zeo-hygtk亚质粒的构建。用ncoi和sphi消化pselect-zeo-hsv1tk,在dntps存在下用t4dnapolymerase进行处理,由此使切割末端平滑化。另一方面,以pcdna3.1/hygro为模板,使用5’-tcaccggtcaccatgaaaaagcctgaactcaccgcg-3’(gt38)(序列号4)及5’-tcaaaggcagaagcaacttctacacagccatcggtcc-3’(gt39)(序列号5),进行利用kod-plus-dnapolymerase(toyobo)的pcr反应(94℃-2分钟→将94℃-15秒钟、58℃-30秒钟、68℃-1分30秒钟进行40个循环→68℃7分钟),得到了约1000碱基的pcr产物。利用in-fusionkit(toyobo)使上述的平滑化pselect-zeo-hsv1tk与1000-basepcr产物连接,得到了pselect-zeo-hygtk(28-10)。
如下所述进行了pmb1kmhygtk供体质粒的构建。以pselect-zeo-hygtk(28-10)作为模板,使用5’-atttaaatcagcggccgcggatctgcgatcgctccg-3’(gt84)(序列号6)及5’-tgtctggccagctagctcaggtttagttggcc-3’(gt85)(序列号7),进行利用kod-plus-dnapolymerase(toyobo)的pcr反应(94℃-2分钟→将94℃-15秒钟、58℃-30秒钟、68℃-3分钟进行了40个循环→68℃7分钟),得到了约2800碱基的pcr产物。利用in-fusionkit(toyobo)使上述的2800-bppcr产物与用noti和nhei消化的pselect-km-hsv1tk(1)连接,得到了pmb1kmhygtk(1)。以后,使用该质粒作为供体质粒载体。
pbs-hprtex2syn(1200)iscei亚质粒的构建
将用于制作本发明中使用的供体质粒载体的亚质粒的构建方法示于以下。在本发明中,“pbs”表示pbluescriptsk+。
如下所述进行了pbs-hprtex2亚质粒的构建。以来自纤维肉瘤(fibrosarcoma)的ht-1080细胞的genomicdna作为模板,使用5’-agcctgggcaacatagcgagacttc-3’(gt28)(序列号8)及5’-tctggtccctacagagtcccactatacc-3’(gt22)(序列号9),进行利用kod-plus-dnapolymerase(toyobo)的pcr反应(94℃-2分钟→将94℃-15秒钟、60℃-30秒钟、68℃-3分30秒钟进行40个循环→68℃7分钟),得到了约2800碱基的pcr产物。以来自纤维肉瘤的ht-1080细胞的genomicdna为模板,使用5’-gctgggattacacgtgtgaaccaacc-3’(gt19)(序列号10)及5’-tggctgcccaatcacctacaggattg-3’(gt24)(序列号11),进行利用kod-plus-dnapolymerase的pcr反应(94℃-2分钟→将94℃-15秒钟、58℃-30秒钟、68℃-8分钟进行40个循环→68℃7分钟),得到了约3100碱基的pcr产物。以上述的2800-bppcr产物和3100-bppcr产物作为模板,使用5’-atccactagttctagaagcctgggcaacatagcgagacttc-3’(gt29)(序列号12)及5’-caccgcggtggcggccgctggctgcccaatcacctacaggattg-3’(gt30)(序列号13),进行利用kod-plus-dnapolymerase(toyobo株式会社,编号kod-101)的pcr反应(94℃-2分钟→将94℃-15秒钟、58℃-30秒钟、68℃-6分钟进行40个循环→68℃7分钟),得到了约5500碱基的pcr产物。利用in-fusionkit(clontechlaboratories,inc.目录号639649)使上述的5500-bppcr产物与用noti消化的pbluescriptsk+连接,得到了pbs-hprtex2(18-7)。以后,使用该质粒作为基因定点突变(site-directedmutagenesis)的模板。
如下所述进行了pbs-hprtex2iscei亚质粒的构建。以来自纤维肉瘤的ht-1080细胞的genomicdna作为模板,使用5’-tagttctagagcggccgcagcctgggcaacatagcgagacttc-3’(gt35)(序列号14)及包含i-scei识别序列(下划线)的5’-attaccctgttatccctaacctggttcatcatcactaatctg-3’(gt34)(序列号15),进行利用kod-plus-dnapolymerase(toyobo株式会社编号kod-101)的pcr反应(94℃-2分钟→将94℃-15秒钟、58℃-30秒钟、68℃-3分钟进行40个循环→68℃7分钟),得到了约2500碱基的pcr产物。以来自纤维肉瘤的ht-1080细胞的genomicdna作为模板,使用包含i-scei识别序列(下划线)的5’-tagggataacagggtaattatgaccttgatttattttgcatacc-3’(gt33)(序列号16)及5’-caccgcggtggcggccgctggctgcccaatcacctacaggattg-3’(gt30)(序列号13),进行利用kod-plus-dnapolymerase的pcr反应(94℃-2分钟→将94℃-15秒钟、58℃-30秒钟、68℃-3分钟进行40个循环→68℃7分钟),得到了约2900碱基的pcr产物。利用in-fusionkit(toyobo)使上述的2500-bppcr产物、2900-bppcr产物、以及用noti消化的pbluescriptsk+连接,得到了pbs-hprtex2iscei(21-1)。以后,使用该质粒作为具有由基因定点突变得到的修饰序列的片段的取代质粒。
如下所述进行了pbs-hprtex2syn亚质粒的构建。以pbs-hprtex2(18-7)作为模板,使用包含修饰后序列syn(下划线)的5’-ggctacgatctcgacctcttttgcatacctaatcattatgc-3’(gt93)(序列号17)及5’-tggttcatcatcactaatctg-3’(gt95)(序列号18),作为基因定点突变法之一,通过使用kod-plus-inversepcrmutagenesiskit(toyobo)得到了pbs-hprtex2syn(inv15)。将用bglii和sphi对pbs-hprtex2syn(inv15)plasmiddna进行消化/凝胶提取而得到的包含syn序列的bglii-sphi片段、与用bglii和sphi消化的hprtex2iscei(21-1)plasmiddna样品进行连接,得到了pbs-hprtex2syn(inv15-2)。
如下所述进行了pbs-hprtex2syn(1200)iscei亚质粒的构建。以pbs-hprtex2syn(inv15-2)作为模板,使用包含iscei序列(下划线)的5’-tagggataacagggtaatattttgtagaaacagggttcgc-3’(gt86)(序列号19)及5’-aaaaatattagctgggagtgg-3’(gt87)(序列号20),并使用kod-plus-inversepcrmutagenesiskit(toyobo),由此得到了pbs-hprtex2syn(1200)iscei(1)。
如下所述进行了pmb1kmhygtk-hprtex2syn(1200)iscei供体质粒载体的构建。进行用noti消化的pbs-hprtex2syn(1200)iscei(1)dna样品与用noti消化的pmb1kmhygtkplasmiddna的连接,得到pmb1kmhygtk-hprtex2syn(1200)iscei(5),确认了插入方向如图2a所示。
[实施例2]使用了供体多核苷酸的基因修饰
(1)
如下所述进行了载体靶向插入体细胞htg786(参照图2b;图3ab)的制作。在电穿孔的前一天将来自纤维肉瘤的ht-1080细胞以3×106细胞10-mldmem培养基/t75烧瓶涂布于6个烧瓶,进行附着培养,在大约24小时后去除培养基,以5mlpbs/t75烧瓶进行添加/去除,以2ml/t75烧瓶添加0.25%胰蛋白酶/1mmedta液,在37℃下温育1分钟,以3ml/t75烧瓶添加dmem培养基,通过3~4次移液(使用10-ml移液管)将细胞剥离/悬浮,回收至1根50-ml试管,进行1,200rpm3分钟的离心,去除上清,添加20mlopti-mem,并通过移液(使用10-ml移液管)将细胞悬浮,在测定细胞数后,再次进行1,200rpm3分钟的离心,去除上清,以1×107~2×107细胞/ml添加opti-mem,将p1000移液管设为800μl,缓慢进行移液,不起泡地重复10~20次,向1.5ml-eppendorf管导入10μg的pmb1kmhygtk-hprtex2syn(1200)iscei供体质粒载体dna(500-1000μg/mlendotoxin-freete)和13.3μg的piscei表达用质粒dna(naturevol.401pp397);500-1000μg/mlendotoxin-freete),导入0.8ml细胞悬浮液并与dna溶液混合,将全部量的细胞dna混合液转移至电穿孔用电击杯(cuvette)(电极间4mm),在冰中放置5分钟,将btx电穿孔仪设定为140mv、脉冲时间70ms、脉冲数3、脉冲间隔200ms,将电击杯安装于装置中,施加电压,将电击杯从装置上取下,在冰中放置5分钟,将全部量的电穿孔液转移至165ml不含选择药物的dmem培养基,缓慢混合,以10ml/直径10-cm盘涂布16个,在37℃、5%co2条件下开始非选择培养(第0天)。在第2天,更换为含100μg/ml潮霉素的dmem培养基,开始选择培养,每隔2、3天用相同的选择培养基进行更换,在第14天以4个盘为代表测定潮霉素抗性细胞集落数,更换为包含100μg/ml潮霉素和7.5μg/ml6-硫鸟嘌呤的dmem培养基,开始双重选择培养,每隔2天或3天用相同的选择培养基进行更换,在第21天对全部盘上的潮霉素/6-硫鸟嘌呤双抗性(htg)细胞集落数进行了测定。
上述实验的结果是,在潮霉素选择下得到了6472个潮霉素抗性细胞集落,在潮霉素/6-硫鸟嘌呤双重选择下得到了14个htg克隆。
如下所述进行了载体靶向插入体分离步骤(参照图2de;参照图3ab)。对于各htg细胞集落,在显微镜下将p1000pipetman设为200μl,剥取细胞,将其悬浮于加入了2ml含有100μg/ml潮霉素和7.5μg/ml6-硫鸟嘌呤的dmem的6孔板中的1个孔中,赋予htg克隆号,在37℃、5%co2条件下进行选择培养,持续培养至汇合附近。去除培养基,以2ml/孔添加/除去pbs,以500μl/孔添加0.25%胰蛋白酶/1mmedta液,在37℃下温育1分钟,以500μl/孔添加dmem培养基,通过3~4次p1000移液将细胞剥离/悬浮,将细胞回收至1根1.5-mleppendorf管,进行2,500rpm3分钟的离心,去除上清,添加100μlpbs,并通过移液将细胞悬浮,分取各40μl至2根cryotubes管,以500μl/cryotubes管中加入cellbanker1plus,保管于-80℃,将剩余的20μlpbs细胞悬浮液进行2,500rpm3分钟离心,去除上清,将细胞团块在-80℃下冷冻保存。在水中将冷冻细胞团块解冻,用geneelutemammaliangenomicdnaminiprepkit(sigma-aldrich,目录号g1n350)提取基因组dna,将其作为100μl样品在4℃下保管。以htg克隆的genomicdna作为模板,使用5’-ttgcaagcagcagattacgc-3’(gt112)(序列号21)及5’-gccactgcacccagccgtatgt-3’(gt69)(序列号22),进行利用kod-fx-dnapolymerase(toyobo株式会社)的pcr反应(94℃-2分钟→将98℃-10秒钟、68℃-8分钟进行40个循环→68℃7分钟),判定了与图2b右同样地产生约7700碱基pcr产物的htg克隆为形成图2a的结构的载体靶向插入体细胞。
上述实验的结果是从14个htg克隆得到了8个载体靶向插入体细胞。这表明,在包含载体的随机插入体和靶向插入体的全部载体基因组插入体的6472个克隆中,8个克隆是载体靶向插入体。因此,获得靶向插入体的实用性为1.2×10-3,这表明可以从1000个载体基因组插入体克隆中得到希望的靶向插入体。
如图3及本说明书中的该说明所示,对于载体靶向插入体中包含修饰前序列(sexa1序列)和修饰后序列(同义转换序列syn)的2种标记序列是否共存、即是否为异型进行了序列分析,结果是确认了在8个载体靶向插入体克隆中,一个为sexa1同型,包含htg786的其它7个为异型或syn同型。异型和syn同型的载体靶向插入体可以进行对基因取代体进行分离的工序(记载于下一段)。
如下所述进行了从载体靶向插入体分离具有基因修饰后序列的基因取代体的步骤(参照图4ab)。使用了作为8个载体靶向插入克隆中的一个的htg786细胞。将该克隆的-80℃保管细胞在37℃水温层中解冻,转移至包含9ml的dmem培养基的50ml试管,进行1,200rpm3分钟离心,去除上清,添加10mldmem培养基,并通过移液使其悬浮,在t75烧瓶中于37℃、5%co2下进行了附着培养。在大约24小时后去除培养基,以10mlpbs/t75烧瓶添加/除去,以1ml/t75烧瓶添加0.25%胰蛋白酶/1mmedta液,在37℃下温育1分钟,以9ml/t75烧瓶添加dmem培养基,通过3~4次移液(使用10-ml移液管)剥离/悬浮细胞,回收至50-ml试管,在细胞数测定后,进行1,200rpm3分钟离心,去除上清,以2×105细胞/ml添加dmem培养基,使该细胞悬浮液50μl(1x104cells)扩散至包含10mldmem培养基的10-cm盘中,在37℃、5%co2下开始附着培养(第0天)。在第5天用包含1μm更昔洛韦(invivogen,目录号#sud-gcv)的dmem培养基更换,开始选择培养。在第7、9、12天进行培养基更换,在第20~25天对更昔洛韦抗性(gcv)细胞集落数进行计数,对于各gcv细胞集落,在显微镜下将p1000pipetman设为200μl,剥取细胞,将其悬浮于加入了2ml含有1μm更昔洛韦的dmem的6孔板中的1个孔中,赋予gcv克隆号,在37℃、5%co2条件下进行选择培养,持续培养至汇合状态。去除培养基,以2ml/孔添加/除去pbs,以500μl/孔添加0.25%胰蛋白酶/1mmedta液,在37℃下温育1分钟,以500μl/孔添加dmem培养基,通过3~4次p1000移液将细胞剥离/悬浮,将细胞回收至1根1.5-mleppendorf管,进行2,500rpm3分钟的离心,去除上清,添加100μlpbs,并通过移液将细胞悬浮,分取各40μl至2根cryotubes管,以500μl/cryotubes管中加入cellbanker1plus,保管于-80℃,将剩余的20μlpbs细胞悬浮液进行2,500rpm3分钟离心,去除上清,将细胞团块在-80℃下冷冻保存。在水中将冷冻细胞团块解冻,用geneelutemammaliangenomicdnaminiprepkit(sigma-aldrich,目录号g1n350)提取基因组dna,将其作为100μl样品在4℃下保管。以gcv克隆的genomicdna作为模板,使用5’-gctgggattacacgtgtgaaccaacc-3’(gt19)(序列号10)及5’-tctggtccctacagagtcccactatacc-3’(gt22)(序列号9),进行利用kod-fx-dnapolymerase(toyobo株式会社编号kfx-101)的pcr反应(94℃-2分钟→将98℃-10秒钟、68℃-1分钟进行40个循环→68℃7分钟),与图4c同样地确认约500碱基的pcr产物,用pcrclean-up(macherey-nagel,740609.250)纯化后,进行了测序。
上述实验的结果是得到12个gcv克隆,且得到了在外显子2上从修饰前序列sexa1(5’-ccaggttatgaccttgatttatttt-3’)(序列号23)被修饰为来自pmb1kmhygtk-hprtex2syn(1200)iscei供体质粒载体的修饰后序列syn(下划线)(5’-ccaggctacgatctcgacctctttt-3’)(序列号24)的克隆。
(2)
再次进行了从载体靶向插入体htg786分离具有基因修饰后序列的基因取代体。将从前面段落中记载的实验(实验1)中得到的12个gcv抗性细胞集落、和从同样的实验(实验2)中得到的9个gcv抗性细胞集落分别以5cellsperwell(96-wellplate)进行极限稀释后,进行培养,对克隆进行纯化,如前面段落中记载的步骤那样,以克隆的genomicdna作为模板,使用5’-gctgggattacacgtgtgaaccaacc-3’(gt19)(序列号10)及5’-tctggtccctacagagtcccactatacc-3’(gt22)(序列号9),进行利用kod-fx-dnapolymerase(toyobo株式会社编号kfx-101)的pcr反应(94℃-2分钟→将98℃-10秒钟、68℃-1分钟进行40个循环→68℃7分钟),与图4c同样地确认约500碱基的pcr产物,用pcrclean-up(macherey-nagel,740609.250)纯化后,进行了序列分析。
基于从这2次实验得到的gcv抗性克隆的序列结果进行分类,总结于下表。
[表1]
这些结果表明,通过将10个左右的gcv抗性细胞集落作为基因取代细胞候选来回收、并进行克隆纯化,可以从gcv抗性克隆获得预期设计的基因取代体(在本试验的情况下为syn同义转换克隆)。
为了最优化供体质粒的结构,构建了供体质粒中的同义转换序列位点与iscei切割位置之间的距离不同的3种供体质粒载体。如实施例1所示,将与包含插入载体骨架的修饰后序列的修饰对象基因座同源的序列接着载体骨架之后的连字符表示为“-hprtiscei(1200)ex2syn”。“ex2syn”表示同义转换修饰后序列syn包含于ex2内,“iscei(1200)”表示在从修饰后序列syn向5’侧1200bp的位置存在i-scei切割酶识别序列。
1)如下所述进行了pbs-hprtiscei(1200)ex2syn亚质粒的构建。以pbs-hprtex2syn(inv15-2)作为模板,使用包含iscei序列(下划线)的5’-tagggataacagggtaatcaaagcactgggattacaagtg-3’(gt117)(序列号25)及5’-ggaggctgagacaggagagttgc-3’(gt118)(序列号26),并使用kod-plus-inversepcrmutagenesiskit(toyobo),由此得到了pbs-hprtiscei(1200)ex2syn(3)。
2)如下所述进行了pbs-hprtiscei(600)ex2syn亚质粒的构建。以pbs-hprtex2syn(inv15-2)作为模板,使用包含iscei序列(下划线)的5’-tagggataacagggtaatcaaagtgctgggattacaggc-3’(gt131)(序列号27)及5’-ggaggccgaggcgggtggatca-3’(gt132)(序列号28),并使用kod-plus-inversepcrmutagenesiskit(toyobo),由此得到了pbs-hprtiscei(600)ex2syn(4)。
3)如下所述进行了pbs-hprtiscei(316)ex2syn亚质粒的构建。以pbs-hprtex2syn(inv15-2)作为模板,使用包含iscei序列(下划线)的5’-tagggataacagggtaattgtatttttagtagagacggg-3’(gt133)(序列号29)及5’-aaaaaattagccgggtgtgg-3’(gt134)(序列号30),并使用kod-plus-inversepcrmutagenesiskit(toyobo),由此得到了pbs-hprtiscei(316)ex2syn(2)。
1)如下所述进行了pmb1kmhygtk-hprtiscei(1200)ex2syn供体质粒载体的构建。进行用noti消化的pbs-hprtiscei(1200)ex2syn(3)dna样品与用noti消化的pmb1kmhygtkplasmiddna的连接,得到pmb1kmhygtk-hprtiscei(1200)ex2syn(1),确认了插入方向与图2a相同。
2)如下所述进行了pmb1kmhygtk-hprtiscei(600)ex2syn供体质粒载体的构建。进行用noti消化的pbs-hprtiscei(600)ex2syn(4)dna样品与用noti消化的pmb1kmhygtkplasmiddna的连接,得到pmb1kmhygtk-hprtiscei(600)ex2syn(1),确认了插入方向与图2a相同。
3)如下所述进行了pmb1kmhygtk-hprtiscei(316)ex2syn供体质粒载体的构建。进行用noti消化的pbs-hprtiscei(316)ex2syn(2)dna样品与用noti消化的pmb1kmhygtkplasmiddna的连接,得到pmb1kmhygtk-hprtiscei(316)ex2syn(1),确认了插入方向与图2a相同。
[实施例3]装载i-scei的仙台病毒载体的构建
为了充分利用仙台病毒载体没有随机插入基因组的危险性的优点,将装载有i-scei序列特异性切割酶基因的仙台病毒载体导入细胞并使其表达,如下所述构建了装载i-scei的仙台病毒载体。
对于以n末端具有核定位信号的i-scei酶的基因,以酿酒酵母(saccharomycescerevisiae)提取dna作为模板,使用5’-ggatcctgcaaagatggataaagcggaattaattcccgagcctccaaaaaagaagagaaaggtcgaattgggtaccatgaaaaatattaaaaaaaatcaagtaatgaatctgggtcc-3’(序列号31)及5’-atgcatttattttaaaaaagtttcggatgaaatagtattaggc-3’(序列号32),进行利用kod-plus-dnapolymerase的pcr反应(94℃-1分钟→将94℃-15秒钟、40℃-gradient-54℃-30秒钟、68℃-1分钟进行30个循环),得到约800碱基的pcr产物,得到了puc-nlsiscei质粒。以该plasmiddna作为模板,使用5’-gtcgacccgggcggccgccatggataaagcggaattaattcccg-3’(gt40)(序列号33)及5’-ctaaagggaagcggccgcttattttaaaaaagtttcgg-3’(gt41)(序列号34),进行利用kod-plus-dnapolymerase的pcr反应(94℃-2分钟→将94℃-15秒钟、55℃-30秒钟、68℃-1分钟进行30个循环→68℃7分钟),得到了约800碱基的pcr产物。利用in-fusionkit(toyobo)使该pcr产物与用noti消化的pci-neo连接,得到了pci-neo-nlsiscei(29-2)。
为了将来自pci-neo-nlsiscei(29-2)的nlsiscei序列特异性切割酶基因装载于仙台病毒载体sev,通过第1pcr得到该nlsiscei序列被分成2部分的第1pcr产物,在第2pcr中,以5’侧的第1pcr产物和3’侧的第1pcr产物作为模板,得到跨越该cas9nuclease序列全长的第3pcr产物,将其装载于仙台病毒载体。在第1pcr中,将该nlsiscei序列分为两部分的原因在于,该nlsiscei序列内存在3个富含a的序列(7a,8a),在富含a的序列上,在仙台病毒载体的生产过程中容易发生由仙台病毒的rna依赖性rna聚合酶导致的错误,因此这是为了避免这样的错误现象。在富含a的序列位点上设定pcr引物,在同义密码子的限制下将各引物序列从a/t取代为g/c。
[表2]
第1pcr的引物和模板dna
使用上排的引物对not1_nls-i-scein_a36g_a78g_a81g_n(别名:nls-i-scein_n1)5’-cgagcctccaaagaagaagagaaaggtcgaattgggtaccatgaaaaatattaagaagaatcaagtaat-3’)(序列号35)、nls-i-scei_a426g_c(5’-gttcgggatggttttcttgttgttaacg-3’)(序列号36)和模板dna,进行利用kod-plus-ver.2-dnapolymerase(toyobo株式会社编号kod-211)的pcr反应(94℃-2分钟→将98℃-10秒钟、55℃-30秒钟、68℃-1分钟进行30个循环→68℃-7分钟),得到了pcr产物#1。使用下排的引物对nls-i-scein_a426g_n(5’-cgttaacaacaagaaaaccatcccgaac-3’)(序列号37)、nls-i-scei_eis_not1_c(5’-atatgcggccgcgatgaactttcaccctaagtttttcttactacggttattttaaaaaagtttcggatg-3’)(序列号38)和模板dna,进行利用kod-fx-dnapolymerase(toyobo株式会社编号kfx-101)的pcr反应(94℃-2分钟→将98℃-10秒钟、68℃-1分钟进行30个循环→68℃-7分钟),得到了pcr产物#2。通过电泳确认pcr产物的大小后,用nucleospintmgelandpcrclean-up(macgerey-nagel目录号740609.250/u0609c)进行了纯化。
[表3]
第2pcr的引物和模板dna
使用在3’端具有引物not1_nls-i-scein_a36g_a78g_a81g_n(别名:nls-i-scein_n1)的5’端29个核苷酸(下划线)的引物not1_nls-i-scein_a36g_n5’-atatgcggccgcgacgccaccatggataaagcggaattaattcccgagcctccaaagaagaagagaaaggtcg-3’(序列号39)、nls-i-scei_eis_not1_c(序列号38)、和模板dna,进行利用kod-fx-dnapolymerase(toyobo株式会社编号kfx-101)的pcr反应(94℃-2分钟→将98℃-10秒钟、68℃-1分钟进行40个循环→68℃-7分钟),得到了全长的pcr产物。通过电泳确认产物大小后,用nucleospintmgelandpcrclean-up(macgerey-nagel目录号740609.250/u0609c)进行了纯化。将用noti消化并凝胶提取出的全长nlsiscei片段与用noti消化并进行了bap处理的质粒psev18+ts15/δfdna(编码缺失f基因、且具有m(g69e/t116a/a183s)、hn(a262t/g264/k461g)、p(d433a/r434a/k437a/l511f)、l(l1361c/l1558i/n1197s/k1795e)突变的仙台病毒载体(wo2003/025570,wo2010/008054)的基因组的dna)进行连接,确认克隆的nlsiscei的碱基序列,得到了装载有对sev最优化的全长nlsiscei的质粒psev18+nlsisceits15/δf。以该质粒dna作为模板,进行仙台病毒再构建,得到了装载nlsiscei的仙台病毒载体sev18+nlsisceits15/δf。插入的序列如下所示。夹在noti位点(下划线)、为了调整6n规则而插入的序列(gac)、kozak序列(双下划线)与eis序列(波浪下划线)、noti位点(下划线)之间的区域是插入序列。该序列包含nlsiscei(序列号41)的编码序列(序列号40的第18-785位)。
[实施例4]供体多核苷酸的载体靶向插入的供体质粒结构的最优化
在使用了实施例2(1)所示的供体多核苷酸的基因修饰法的电穿孔时,使用pci-neo-nlsiscei(29-2)dna(12.6μg)代替i-scei表达用质粒dna(naturevol.401pp397),利用3种供体质粒分别进行了载体靶向插入体的制作。
[表4]
piscei:i-scei表达用质粒(naturevol.401pp397)
leftarm:iscei切割位点的左侧同源区域长度(bp)(参照图1a);syn同义转换序列位点的左侧同源区域长度(bp)(参照图2a)
rightarm:syn同义转换序列位点的右侧同源区域长度(bp)(参照图1a参照);iscei切割位点的右侧同源区域长度(bp)(参照图2a)
这些试验如实施例2(1)所示那样使用dmem/10%fbs培养基来进行。在使用了iscei(600)syn型供体质粒的第3次试验中,使用了dmem/2%fbs/1/100容量glutamax-1(100x)(gibco、产品编号35050-061)培养基。在该低fbs条件下,用50μg/ml潮霉素、0.94μg/ml6-硫鸟嘌呤进行了选择培养。
根据这些结果可知,切割点外侧同源区域长度、设计序列外侧同源区域长度、切割点与设计序列的距离没有限定,优选外侧同源区域长度为1960bp以上,切割点与设计序列的距离为316bp以上。
[实施例5]利用装载nlsiscei的仙台病毒载体进行的供体多核苷酸的载体靶向插入
在感染装载nlsiscei的仙台病毒载体的前一天,以5×105个ht-1080细胞/2-mldmem/10%fbs培养基/孔涂布6孔板,进行附着培养,在大约24小时后去除培养基,添加2mlopti-mem,放置于37℃,另一方面,测定1孔内的细胞数,计算出感染的全部细胞数,采集相当于感染复数3的装载nlsiscei的仙台病毒载体sev18+nlsisceits15/δf(实施例3),通过用opti-mem稀释,调整为0.5mlsev-nlsiscei溶液/孔,去除预先添加的opti-mem,加入0.5mlsev-nlsiscei溶液/孔,在32℃、5%co2条件下开始吸附感染,每15分钟进行混合操作,在2小时后去除sev-cas9溶液,添加2mlopti-mem/孔,以2-mldmem培养基(含6-硫鸟嘌呤)/孔进行更换,在32℃、5%co2条件下进行培养。大约在24小时后更换新的dmem培养基,转移至35℃、5%co2,保持该状态培养大约24小时。
接着,回收细胞,与实施例2(1)同样地进行记载于下表的用于导入供体质粒的电穿孔的操作、细胞涂布、非选择培养和选择培养,载体靶向体分离的操作也与实施例2(1)同样地进行。
[表5]
sev/ts15:sev18+nlsisceits15/δf仙台病毒载体
在2次试验中,第1次按照上述的条件(使用dmem/10%fbs培养基,仙台病毒载体的吸附感染32℃-24小时、35℃-24小时,细胞涂布后的非选择培养37℃-3天及选择培养37℃)进行。第2次使用dmem/2%fbs/1/100容量glutamax-1(100x)(gibco、产品编号35050-061)培养基,在仙台病毒载体的吸附感染32℃-48小时、细胞涂布后的非选择培养35℃-3天及选择培养37℃的条件下进行。在该低fbs条件下,用50μg/ml潮霉素、0.94μg/ml6-硫鸟嘌呤进行了选择培养。
根据这些结果可知,用于细胞内切割的切割酶基因表达载体不限定于质粒型、病毒载体型中的任一种,为了避免切割酶基因表达载体的随机插入,优选为该病毒载体型。
[实施例6]利用在细胞外实施了同源区域内切割的供体质粒进行的供体多核苷酸的载体靶向插入
通过使用了实施例2(1)所示的供体多核苷酸的基因修饰法的电穿孔,仅导入预先进行了切割的供体质粒(10μg),进行了载体靶向插入体的制作。
[表6]
leftarm:xmai或iscei切割位点的左侧同源区域长度(bp)(参照图1a);syn同义转换序列位点的左侧同源区域长度(bp)(参照图2a)
rightarm:syn同义转换序列位点的右侧同源区域长度(bp)(参照图1a);xmai或iscei切割位点的右侧同源区域长度(bp)(参照图2a)
根据这些结果,确认了供体质粒的同源区域内切割可以是细胞外或细胞内的任一者。但是,由于细胞内同源区域内切割基本上不受到切割点外侧同源区域长度、设计序列外侧同源区域长度、切割点与设计序列的距离的影响,因此优选为细胞内同源区域内切割。
对本发明的优选方式进行了详细说明,对于本领域技术人员而言,可以对这些方式进行变更是显而易见的。因此,本发明包括可以通过除本说明书中详细记载以外的方法、方式来实施本发明。即,本发明包括与附带的“权利要求书”的思想或其本质部分相同范围所包含的所有变更。
工业实用性
根据本发明,提供通过包含阳性选择标记基因及阴性选择标记基因的多核苷酸连接包含可切割位点的基因组片段的两端而成的新型供体多核苷酸。通过使用该供体多核苷酸,可以在供体多核苷酸的同源位点进行切割而不在靶基因座内进行切割,从而避免被称为off-target的在除靶序列以外导入突变的可能性,可以仅对靶基因进行修饰。本发明是对基因序列进行精密修饰的分子遗传学技术,作为以基因治疗、品种改良、生物技术创造为目的而对基因序列进行精密修饰的分子遗传学系统是有用的。
序列表
<110>株式会社爱医隆集团(i'romgroupco.,ltd.)
<120>用于修饰靶序列的多核苷酸及其用途
<130>d4-a1701p
<150>jp2017-141691
<151>2017-07-21
<160>42
<170>patentinversion3.5
<210>1
<211>18
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>1
tagggataacagggtaat18
<210>2
<211>32
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>2
cttaattaacctgcagccggaattgccagctg32
<210>3
<211>35
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>3
atgtggtatggaattcggtggccctcctcacgtgc35
<210>4
<211>36
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>4
tcaccggtcaccatgaaaaagcctgaactcaccgcg36
<210>5
<211>37
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>5
tcaaaggcagaagcaacttctacacagccatcggtcc37
<210>6
<211>36
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>6
atttaaatcagcggccgcggatctgcgatcgctccg36
<210>7
<211>32
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>7
tgtctggccagctagctcaggtttagttggcc32
<210>8
<211>25
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>8
agcctgggcaacatagcgagacttc25
<210>9
<211>28
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>9
tctggtccctacagagtcccactatacc28
<210>10
<211>26
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>10
gctgggattacacgtgtgaaccaacc26
<210>11
<211>26
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>11
tggctgcccaatcacctacaggattg26
<210>12
<211>41
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>12
atccactagttctagaagcctgggcaacatagcgagacttc41
<210>13
<211>44
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>13
caccgcggtggcggccgctggctgcccaatcacctacaggattg44
<210>14
<211>43
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>14
tagttctagagcggccgcagcctgggcaacatagcgagacttc43
<210>15
<211>42
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>15
attaccctgttatccctaacctggttcatcatcactaatctg42
<210>16
<211>44
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>16
tagggataacagggtaattatgaccttgatttattttgcatacc44
<210>17
<211>41
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>17
ggctacgatctcgacctcttttgcatacctaatcattatgc41
<210>18
<211>21
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>18
tggttcatcatcactaatctg21
<210>19
<211>40
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>19
tagggataacagggtaatattttgtagaaacagggttcgc40
<210>20
<211>21
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>20
aaaaatattagctgggagtgg21
<210>21
<211>20
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>21
ttgcaagcagcagattacgc20
<210>22
<211>22
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>22
gccactgcacccagccgtatgt22
<210>23
<211>25
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>23
ccaggttatgaccttgatttatttt25
<210>24
<211>25
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>24
ccaggctacgatctcgacctctttt25
<210>25
<211>40
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>25
tagggataacagggtaatcaaagcactgggattacaagtg40
<210>26
<211>23
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>26
ggaggctgagacaggagagttgc23
<210>27
<211>39
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>27
tagggataacagggtaatcaaagtgctgggattacaggc39
<210>28
<211>22
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>28
ggaggccgaggcgggtggatca22
<210>29
<211>39
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>29
tagggataacagggtaattgtatttttagtagagacggg39
<210>30
<211>20
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>30
aaaaaattagccgggtgtgg20
<210>31
<211>44
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>31
gtcgacccgggcggccgccatggataaagcggaattaattcccg44
<210>32
<211>38
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>32
ctaaagggaagcggccgcttattttaaaaaagtttcgg38
<210>33
<211>44
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>33
gtcgacccgggcggccgccatggataaagcggaattaattcccg44
<210>34
<211>38
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>34
ctaaagggaagcggccgcttattttaaaaaagtttcgg38
<210>35
<211>69
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>35
cgagcctccaaagaagaagagaaaggtcgaattgggtaccatgaaaaatattaagaagaa60
tcaagtaat69
<210>36
<211>28
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>36
gttcgggatggttttcttgttgttaacg28
<210>37
<211>28
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>37
cgttaacaacaagaaaaccatcccgaac28
<210>38
<211>69
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>38
atatgcggccgcgatgaactttcaccctaagtttttcttactacggttattttaaaaaag60
tttcggatg69
<210>39
<211>73
<212>dna
<213>人工序列
<220>
<223>人工序列
<400>39
atatgcggccgcgacgccaccatggataaagcggaattaattcccgagcctccaaagaag60
aagagaaaggtcg73
<210>40
<211>830
<212>dna
<213>人工序列
<220>
<223>人工序列
<220>
<221>cds
<222>(18)..(785)
<400>40
gcggccgcgacgccaccatggataaagcggaattaattcccgagcctcca50
metasplysalagluleuileproglupropro
1510
aagaagaagagaaaggtcgaattgggtaccatgaaaaatattaagaag98
lyslyslysarglysvalgluleuglythrmetlysasnilelyslys
152025
aatcaagtaatgaatctgggtccgaactctaaactgctgaaagaatac146
asnglnvalmetasnleuglyproasnserlysleuleulysglutyr
303540
aaatcccagctgatcgaactgaacatcgaacagttcgaagcaggtatc194
lysserglnleuilegluleuasnilegluglnpheglualaglyile
455055
ggtctgatcctgggtgatgcttacatccgttctcgtgatgaaggtaaa242
glyleuileleuglyaspalatyrileargserargaspgluglylys
60657075
acctactgtatgcagttcgagtggaaaaacaaagcatacatggaccac290
thrtyrcysmetglnpheglutrplysasnlysalatyrmetasphis
808590
gtatgtctgctgtacgatcagtgggtactgtccccgccgcacaaaaaa338
valcysleuleutyraspglntrpvalleuserproprohislyslys
95100105
gaacgtgttaaccacctgggtaacctggtaatcacctggggcgcccag386
gluargvalasnhisleuglyasnleuvalilethrtrpglyalagln
110115120
actttcaaacaccaagctttcaacaaactggctaacctgttcatcgtt434
thrphelyshisglnalapheasnlysleualaasnleupheileval
125130135
aacaacaagaaaaccatcccgaacaacctggttgaaaactacctgacc482
asnasnlyslysthrileproasnasnleuvalgluasntyrleuthr
140145150155
ccgatgtctctggcatactggttcatggatgatggtggtaaatgggat530
prometserleualatyrtrpphemetaspaspglyglylystrpasp
160165170
tacaacaaaaactctaccaacaaatcgatcgtactgaacacccagtct578
tyrasnlysasnserthrasnlysserilevalleuasnthrglnser
175180185
ttcactttcgaagaagtagaatacctggttaagggtctgcgtaacaaa626
phethrpheglugluvalglutyrleuvallysglyleuargasnlys
190195200
ttccaactgaactgttacgtaaaaatcaacaaaaacaaaccgatcatc674
pheglnleuasncystyrvallysileasnlysasnlysproileile
205210215
tacatcgattctatgtcttacctgatcttctacaacctgatcaaaccg722
tyrileaspsermetsertyrleuilephetyrasnleuilelyspro
220225230235
tacctgatcccgcagatgatgtacaaactgcctaatactatttcatcc770
tyrleuileproglnmetmettyrlysleuproasnthrileserser
240245250
gaaacttttttaaaataaccgtagtaagaaaaacttagggtgaaagttcatcgcg825
gluthrpheleulys
255
gccgc830
<210>41
<211>256
<212>prt
<213>人工序列
<220>
<223>合成的结构体
<400>41
metasplysalagluleuileprogluproprolyslyslysarglys
151015
valgluleuglythrmetlysasnilelyslysasnglnvalmetasn
202530
leuglyproasnserlysleuleulysglutyrlysserglnleuile
354045
gluleuasnilegluglnpheglualaglyileglyleuileleugly
505560
aspalatyrileargserargaspgluglylysthrtyrcysmetgln
65707580
pheglutrplysasnlysalatyrmetasphisvalcysleuleutyr
859095
aspglntrpvalleuserproprohislyslysgluargvalasnhis
100105110
leuglyasnleuvalilethrtrpglyalaglnthrphelyshisgln
115120125
alapheasnlysleualaasnleupheilevalasnasnlyslysthr
130135140
ileproasnasnleuvalgluasntyrleuthrprometserleuala
145150155160
tyrtrpphemetaspaspglyglylystrpasptyrasnlysasnser
165170175
thrasnlysserilevalleuasnthrglnserphethrphegluglu
180185190
valglutyrleuvallysglyleuargasnlyspheglnleuasncys
195200205
tyrvallysileasnlysasnlysproileiletyrileaspsermet
210215220
sertyrleuilephetyrasnleuilelysprotyrleuileprogln
225230235240
metmettyrlysleuproasnthrilesersergluthrpheleulys
245250255
<210>42
<211>21
<212>prt
<213>人工序列
<220>
<223>人工序列
<400>42
metasplysalagluleuileprogluproprolyslyslysarglys
151015
valgluleuglythr
20
- 还没有人留言评论。精彩留言会获得点赞!