一种光学纯氯前列醇的合成方法与流程
本发明涉及医药制备技术领域,特别涉及一种光学纯氯前列醇的合成方法。
背景技术:
前列腺素(prostaglandins,简称pgs)是一类具有广泛生理活性的重要内源性物质。其各化合物均具有相同的母体结构,主要区别是和母体结构相连的上、下侧链结构的不同。氯前列醇,英文名称为:cloprostenol,分子式为:c22h28clo6。2003年10月,农业部批准注射用氯前列醇及其钠盐为新兽药。《中国兽药典》收载的氯前列醇为混旋化合物,混旋化合物中左旋和右旋化合物结构上的区别是下侧链羟基手性构型差异。
氯前列醇具有强大溶解黄体和直接兴奋子宫平滑肌作用,主要用于治疗持久黄体、黄体囊肿而引起的不发情;促进猪、牛、羊的同期发情和发情调控;诱导兽类的引产、流产等分娩控制;母畜产后护理、产后子宫内膜炎防治及促进卵巢、子宫修复等。
cn104513186a公开了一种光学纯的右旋氯前列醇的制备方法,公告日为2016年10月05日。其以2,5-降冰片二烯为原料,经过十八步反应制得,该方法步骤冗长,反应收率非常低;同时,手性拆分还要经过三次重结晶,反应中间体需要柱层析纯化,并不适合工业化放大生产。
《chirality》【27:392–396(2015)】报道了改进的右旋氯前列醇的合成方法,该方法以联苯保护的左旋科里内酯为原料,经过九步反应,总收率为25.4%。
因此,现有的技术中右旋氯前列醇的制备方法均存在制备复杂,收率低的问题。
技术实现要素:
为解决上述背景技术中右旋氯前列醇的制备方法复杂,收率低的问题,本发明提供一种光学纯氯前列醇的合成方法,包括以下步骤:
步骤a、将左旋科里内酯二醇和氧化剂加入溶剂中,并在dmn-azado催化剂作用下,进行选择性氧化反应,进行淬灭、萃取、洗涤、干燥、过滤,得到含醛基的科里内酯单醇,即为化合物i;
具体地,进行选择性氧化反应后,加入饱和na2s2o3溶液进行淬灭,水相使用二氯甲烷萃取,合并有机相再使用饱和氯化钠溶液洗涤、无水硫酸钠干燥、过滤,制得化合物i的二氯甲烷溶液,用于步骤b使用。
步骤b、将化合物i、膦酸酯与溶剂在氢氧化锂的催化下进行烯烃化反应,经过淬灭、萃取、洗涤、过滤、浓缩、干燥,得到化合物ii;
具体地,进行烯烃化反应结束后,加入饱和氯化钠溶液淬灭,水相再使用乙酸乙酯萃取;合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩后得棕色油状物,再加入甲醇溶液,进行搅拌,降温至0℃后,恒温搅拌1h,过滤,滤饼再使用冷的甲醇淋洗、干燥,即得到化合物ii。
步骤c、以四氢呋喃为溶剂,采用二异丁基氢化铝将化合物ii进行还原反应,经过淬灭、萃取、洗涤、干燥、过滤、浓缩,得到中间体化合物iii;
具体地,进行还原反应后,加入甲醇进行淬灭,反应温度不高于0℃,再于室温下反应,加入盐酸溶液搅拌至澄清;静置分层后,水相再使用乙酯乙酯萃取,合并有机相,分别使用饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩,得白色固体,即得到中间体化合物iii。
步骤d、将中间体化合物iii进行wittig反应,经萃取、洗涤、干燥、过滤、浓缩得到化合物iv;
具体地,wittig反应后,加入饱和氯化钠溶液,静置分层,水相再使用乙酸乙酯萃取,合并有机相,再使用氢氧化钠溶液洗涤,静置分层后,有机相弃去;合并所有水相,利用磷酸酸化再使用乙酸乙酯萃取,合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩后得淡黄色液体,再加入乙酸乙脂,搅拌均匀后冷冻析晶,即得到化合物iv。
步骤e、将化合物iv溶解于溶剂中,加入(s)-苯乙胺进行反应,再进行重结晶,得到化合物v;
具体地,以(s)-苯乙胺为手性诱导剂对化合物iv进行手性拆分。
步骤f、将化合物v进行酸化调节后,进行萃取、浓缩、析晶、干燥,即得到光学纯氯前列醇;
具体地,酸化调节后,使用乙酸乙酯萃取,合并后的有机相再使用饱和氯化钠洗涤、无水硫酸钠干燥、过滤,浓缩后得淡黄色固体;再将所述淡黄色固体转移至乙酸乙酯/石油醚混合液中,并加热溶解,再冷却析晶,过滤、洗涤、干燥,得光学纯的氯前列醇。
进一步地,步骤a中,所述dmn-azado催化剂的使用量为1-10mol%;所述溶剂为二氯甲烷、饱和碳酸氢钠或乙腈中的一种或至少两种混合,使用量为反应底物的1-10l/mol;反应温度优选为0-30℃。
进一步地,步骤a中,所述氧化剂为碘苯乙酸酯、次氯酸钠、氯酸钠、溴化钾、四丁基溴化铵中的一种或多种混合。
进一步地,步骤b中,所述溶剂为乙腈、四氢呋喃或者二氯甲烷中的一种或至少两种混合,使用量为反应底物的1-10l/mol;所述氢氧化锂的使用当量为反应底物的1.5-3倍;所述烯烃化反应的温度为10-30℃。
进一步地,步骤c中,所述四氢呋喃使用体积为底物体积的5-20倍,dibal-h使用当量为底物的4-10倍摩尔当量;反应温度为-80至-50℃之间。
进一步地,步骤d中,所述wittig反应过程包括:将(4-羧丁基)三苯基溴化膦、hmpa和四氢呋喃,与nahmds反应生成wittig盐,然后再将化合物iii的四氢呋喃溶液加入至反应体系,即可通过wittig反应得到消旋体的目标化合物iv。
进一步地,步骤e中,所述溶剂为甲醇和丙酮的混合溶液;其中,甲醇:丙酮:化合物iv:(s)-苯乙胺=0.5-1ml:3-6ml:1g:0.1424g;
加入(s)-苯乙胺后,加热使溶液变澄清,然后降温搅拌析晶,再将体系温度降至-10℃以下,搅拌析晶;晶体过滤后,使用甲醇和丙酮的混合液淋洗、过滤、干燥,得白色固体。
所述重结晶过程包括:将所述白色固体溶解于甲醇和丙酮的混合溶剂中,搅拌加热溶解,再降温至-10℃以下,搅拌、过滤、干燥,即完成重结晶,并得到化合物v。
进一步地,步骤f中,采用磷酸进行酸化调节,调节ph=3-4。
本发明以简单易得的左旋科里内酯二醇为原料,经过六步反应,制得右旋氯前列醇。与现有的合成方法相比具有明显的优势:
1、直接以廉价易得的左旋科里内酯二醇为原料,无需要对羟基进行选择性保护,而是直接选择性氧化伯醇,进行合成反应;
2、反应步骤短,目标产物光学纯度高,且反应总收率为33.6%,高于现有的生产工艺;
3、各步均为常见的合成方法,操作方便、反应后处理简单、无需柱层析纯化,可实现工业化批量生产。
本发明公开了一种以商品化左旋科里内酯二醇为原料,低成本合成光学纯氯前列醇的新方法。该工艺具有原料易得、步骤短、操作简单和对设备要求低等特点;同时中间体纯化方法简单、杂质容易控制、产品收率高等特性。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
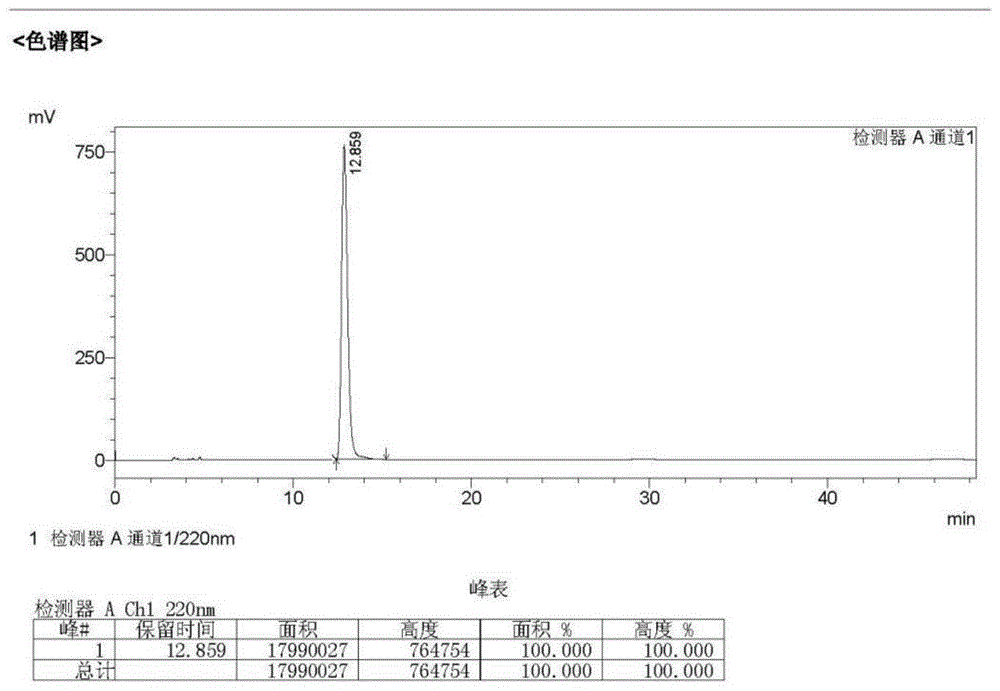
图1为本发明提供的右旋氯前列醇的色谱图;
图2为本发明提供的混旋氯前列醇的色谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供一种光学纯氯前列醇的合成方法,包括以下步骤:
步骤a、将左旋科里内酯二醇和氧化剂加入溶剂中,并在dmn-azado催化剂作用下,进行选择性氧化反应,进行淬灭、萃取、洗涤、干燥、过滤,得到含醛基的科里内酯单醇,即为化合物i;
工艺路线如下:
具体地,进行选择性氧化反应后,加入饱和na2s2o3溶液进行淬灭,水相使用二氯甲烷萃取,合并有机相再使用饱和氯化钠溶液洗涤、无水硫酸钠干燥、过滤,制得化合i的二氯甲烷溶液,用于步骤b使用。
步骤b、将化合物i、膦酸酯与溶剂在氢氧化锂的催化下进行烯烃化反应,经过淬灭、萃取、洗涤、过滤、浓缩、干燥,得到化合物ii;
工艺路线如下:
具体地,进行烯烃化反应结束后,加入饱和氯化钠溶液淬灭,水相再使用乙酸乙酯萃取;合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩后得棕色油状物,再加入甲醇溶液,进行搅拌,降温至0℃,恒温搅拌1h,过滤,滤饼再使用冷的甲醇淋洗、干燥,即得到化合物ii。
步骤c、以四氢呋喃为溶剂,采用二异丁基氢化铝将化合物ii进行还原反应,经过淬灭、萃取、洗涤、干燥、过滤、浓缩,得到中间体化合物iii;
工艺路线如下:
具体地,进行还原反应后,加入甲醇进行淬灭,反应温度不高于0℃,再于室温下反应,加入盐酸溶液搅拌至澄清;置分层后,水相再使用乙酯乙酯萃取,合并有机相,分别使用饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩,得白色固体,即得到中间体化合物iii。
步骤d、将中间体化合物iii进行wittig反应,经萃取、洗涤、干燥、过滤、浓缩得到消旋体的目标化合物iv;
工艺路线如下:
具体地,wittig反应后,加入饱和氯化钠溶液,静置分层,水相再使用乙酸乙酯萃取,合并有机相,再使用氢氧化钠溶液洗涤,静置分层后,有机相弃去;合并所有水相,利用15%磷酸酸化至ph=2,再使用乙酸乙酯萃取,合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩后得淡黄色液体,再加入乙酸乙脂,搅拌均匀后冷冻析晶,即得到化合物iv。
步骤e、将化合物iv溶解于溶剂中,加入(s)-苯乙胺进行反应,再进行重结晶,得到化合物v;
工艺路线如下:
步骤f、将化合物v进行酸化调节后,进行萃取、浓缩、析晶、干燥,即得到光学纯氯前列醇;
工艺路线如下:
具体地,酸化调节后,使用乙酸乙酯萃取,合并后的有机相再使用饱和氯化钠洗涤、无水硫酸钠干燥、过滤,浓缩后得淡黄色固体;再将所述淡黄色固体转移至乙酸乙酯/石油醚混合液中,并加热溶解,再冷却析晶,过滤、洗涤、干燥,得光学纯的氯前列醇。
本发明提供的光学纯氯前列醇的合成方法整体工艺路线如下:
优选地,步骤a中,所述dmn-azado催化剂的使用量为1-10mol%;所述溶剂为二氯甲烷、饱和碳酸氢钠或乙腈中的一种或至少两种混合,使用量为反应底物的1-10l/mol。
优选地,步骤a中,所述氧化剂为碘苯乙酸酯、次氯酸钠、氯酸钠、溴化钾、四丁基溴化铵中的一种或多种混合。
优选地,步骤b中,所述溶剂为乙腈、四氢呋喃或者二氯甲烷中的一种或至少两种混合,使用量为反应底物的1-10l/mol;所述氢氧化锂的使用当量为反应底物的1.5-3倍;所述烯烃化反应的温度为10-30℃。
优选地,步骤c中,所述四氢呋喃使用体积为底物体积的5-20倍,dibal-h使用当量为底物的4-10倍摩尔当量;反应温度为-80至-50℃之间。
优选地,步骤d中,所述wittig反应过程包括:将(4-羧丁基)三苯基溴化膦、hmpa和四氢呋喃,与nahmds反应生成wittig盐,然后再将化合物iii的四氢呋喃溶液加入至反应体系,即可通过wittig反应得到消旋体的目标化合物iv。
优选地,步骤e中,所述溶剂为甲醇和丙酮的混合溶液;
加入(s)-苯乙胺后,加热使溶液变澄清,然后降温搅拌析晶,再将体系温度降至-10℃以下,搅拌析晶;晶体过滤后,使用甲醇和丙酮的混合液淋洗、过滤、干燥,得白色固体;
所述重结晶过程包括:将所述白色固体溶解于甲醇和丙酮的混合溶剂中,搅拌加热溶解,再降温至-10℃以下,搅拌、过滤、干燥,即完成重结晶,并得到化合物v。
优选地,步骤f中,采用磷酸进行酸化调节,调节ph=3-4。
本发明还提供以下具体制备实施例:
(1)制备化合物i
在装有恒压滴液漏斗、温度计及机械搅拌的3l三口瓶中分别加入左旋科里内酯二醇(100g,0.5814mol),dmn-azado(0.986g,0.0058mol)、溴化钾kbr(6.918g,0.058mol)、四丁基溴化铵(n-bu4nbr,9.56g,0.029mol)、二氯甲烷(1l)及饱和碳酸氢钠溶液(100ml)。
然后将上述混合体系,冷却至内温≤0℃,并恒温搅拌10min,将naocl(0.70mol)溶液缓慢滴加至反应体系,控制反应温度不高于5℃。滴加结束,再恒温搅拌30min。反应结束后,体系中加入饱和na2s2o3(200ml)淬灭反应,水相使用二氯甲烷萃取(0.2l*2)。合并有机相再使用饱和氯化钠溶液洗涤、无水硫酸钠干燥、过滤,制得化合物i的二氯甲烷溶液,直接应用于下步反应。
(2)制备化合物ii
在装有温度计、机械搅拌及滴液漏斗的3l的反应釜中,顺次加入lioh˙h2o(58.6g,1.40mol)和乙腈(400ml)。开启搅拌,室温搅拌5min。接着将下侧链中间体膦酸酯(254g,0.87mol),缓慢滴加至上述体系,10min滴完,再室温搅拌1h。然后再将上述反应中得到的中间化合物i的二氯甲烷溶液,缓慢的滴加至反应体系,滴加结束,室温搅拌1h。hplc监控反应结束后,加入饱和氯化钠溶液1l淬灭反应,水相再使用乙酸乙酯萃取(500ml*2)。合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩后得棕色油状物251g,再加入1000ml的甲醇溶液,开启搅拌,降温至0℃,有大量白色固体析出。在该温度下恒温搅拌1h,过滤,滤饼再使用300ml冷的甲醇淋洗、干燥,得化合物ii(181g,纯度98%),白色固体,两步收率92%。
(3)制备化合物iii
在10l的反应釜中,化合物ii(181g,0.539mol)及无水thf(3.6l)。将反应釜内温度降至-75℃,缓慢的滴加1.5mol/l的dibal-h甲苯溶液(1.44l,2.156mol),控制加入速度,维持反应温度-75至-60℃。滴加结束后,再恒温反应1h,接着将反应温度提高至-40℃,再恒温反应1h。hplc检测反应结束后,低温下缓慢的滴加甲醇淬灭反应,控制滴加速度,使反应温度不高于0℃。滴加结束,再室温反应1h,此时体系有大量胶状物析出。将配制好的4mol/l盐酸溶液1500ml,加入至上述体系,搅拌至体系变澄清。静置分层后,水相再使用乙酯乙酯萃取(1l*3),合并有机相,分别使用饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩,得白色固体168g,收率92%,即为化合物iii。
(4)制备化合物iv
在装有温度计、机械搅拌及滴液漏斗的20l的反应釜中,顺次加入上侧链中间体(4-羧丁基)三苯基溴化膦(1318g,2.97mol)、hmpa(100ml)和四氢呋喃(6000ml)。开启搅拌,将反应釜内温度降至-40℃,再缓慢的将nahmds(5.94mol,2.97l)滴加至反应体系,并恒温反应30min,接着再升温至-25℃,搅拌1h。然后再将化合物iii(181g,0.495mol),溶解于800ml的四氢呋喃中,缓慢的滴加至反应体系,滴加结束,再恒温搅拌1h。然后缓慢回温至室温,搅拌过夜。反应结束后,缓慢的加入饱和氯化钠溶液4l,静置分层,水相再使用乙酸乙酯萃取(1l*3),合并有机相,再使用1mol/l的氢氧化钠溶液洗涤,静置分层后,有机相弃去。合并所有水相,利用15%磷酸酸化至ph=2,再使用乙酸乙酯萃取(1000ml*4),合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、过滤、浓缩后得淡黄色液体512g。再加入3l乙酸乙脂,搅拌均匀后冷冻析晶。得粗品化合物iv208g,收率91%。
(5)制备化合物iv
将化合物iv(208g,0.49mol),溶解于装有1200ml的甲醇和4800ml丙酮的溶液的析晶釜中,室温搅拌10min。然后将(s)-苯乙胺(30.2g,0.25mol)缓慢的滴加至上述体系,加热至40℃,使溶液变澄清,然后降温至10℃,搅拌析晶2h。然后再将反应体系的温度降至-10℃,搅拌析晶12h。将晶体过滤后,滤饼再使用混合液(500ml,1:4)淋洗、过滤、干燥,得白色固体135g。将上述固体与400ml的甲醇和1600ml的丙酮混合,搅拌加热至40℃溶解,再以5℃/h的速率降温至-10℃,搅拌12h。过滤、干燥得白色固体化合物v130g,收率97%。
(6)光学纯氯前列醇的制备
将化合物v(130g,0.238mol),溶解于装有1200ml的水中,再使用85%的磷酸调节ph=3-4,室温搅拌30min。然后再使用乙酸乙酯萃取(400ml*4),合并后的有机相再使用饱和氯化钠洗涤、无水硫酸钠干燥、过滤,浓缩后得淡黄色固体105g。再将该固体转移至1000ml含75%的乙酸乙酯/石油醚混合液中,并加热至40℃溶解,再冷却至-10℃析晶12h。过滤、洗涤、干燥,得光学纯的氯前列醇98.6g,纯度98.1%,ee值100%。
将上述制备的相关样品进行色谱分析,其中:图2为中间体化合物iii,即混旋氯前列醇的色谱图,前峰为右旋,后峰为左旋;图1为拆分后得到的右旋结构,即制得的光学纯的氯前列醇;由二图对比可知,图1中没有含有17.47min的峰,可知得到的右旋结构纯度高,达到了光学醇。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!