一种炔基功能化共价三嗪聚合物、其制备方法及应用与流程
本发明属于有机半导体材料技术领域,尤其涉及一种炔基功能化共价三嗪聚合物、其制备方法及应用。
背景技术:
过氧化氢是一种绿色的氧化剂,常常被应用于化学合成,污水处理,杀菌,漂白等,工业上是通过蒽醌法生产过氧化氢,这种方法耗能大而且会排出对动植物有害的副产物。因此寻求一种廉价、耗能低、且环境友好的新型合成过氧化氢的方法一直是该领域的研究热点,而利用太阳能(可见光)催化氧气和水反应生成过氧化氢的方法被研究者视为最有前景的解决方案而被重点研究。
现有光催化产过氧化氢材料中一类是二氧化钛、钒酸铋、氧化亚铜等金属氧化物半导体,这些材料虽然已经进行了较深入的研究且在光催化制过氧化氢中表现出较好的性能,但是由于它们含有金属因而增加了催化剂的合成成本,并且它们的带隙不可控导致其催化制过氧化氢的选择性较差。另一类是基于氮化碳的有机半导体,这类半导体合成成本廉价,带隙可控,而且光催化制过氧化氢的选择性较高,但是在催化体系中常常需要加入空穴牺牲剂,如甲醇,乙醇,三乙醇胺等等,因而不利于实际的应用。
共价三嗪有机框架是一种结构中含有三嗪环的聚合物有机半导体材料,其能带结构具备一定的可控性,由于其在可见光下具有较好的吸收因而很多三嗪类材料都被成功地应用于光解水。德国《应用化学》(angewandtechemieinternationaledition,2008年第47卷第3450页)报道了一种离子热法合成共价三嗪框架的方法,但该法需要400℃的高温,而且反应时间很长,大约40小时,导致该法制备的材料混有高度碳化的杂质,最后得到的材料都是大块状形貌很难进行二维剥离,带隙过窄也不利于进一步的光催化应用。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种炔基功能化共价三嗪聚合物、其制备方法及应用,该方法制备的炔基功能化共价三嗪聚合物易剥离形成二维纳米片层材料,且在可见光范围内具有较好的吸收,可在可见光照射下催化氧气还原和水氧化生成过氧化氢。
本发明提供了一种炔基功能化共价三嗪聚合物的制备方法,包括:
s1)将式(i)所示的对称双氰基有机小分子在保护气氛中、三氟甲磺酸的催化下进行环三聚反应,得到炔基功能化共价三嗪聚合物;
其中,n为1~5的整数。
优选的,所述步骤s1)具体为:
将式(i)所示的对称双氰基有机小分子与三氟甲磺酸在保护气氛中、低温条件下混合反应,然后转移至室温进行反应,最后进行热处理,得到炔基功能化共价三嗪聚合物。
优选的,所述低温条件为0℃~-10℃;混合反应的时间为10~30min;转移至室温进行反应的时间为60~120min;所述热处理的温度为50℃~80℃;所述热处理的时间为20~60min。
优选的,所述n为1或2。
优选的,所述式(i)所示的对称双氰基有机小分子与三氟甲磺酸的比例为(10~300)mg:1ml。
优选的,还包括:
将所述炔基功能化共价三嗪聚合物分散于有机溶剂与氨水的混合溶液中,超声处理,离心后,得到炔基功能化共价三嗪聚合物的二维纳米片。
优选的,所述有机溶剂与氨水的体积比为(2~10):1;所述有机溶剂为乙腈;所述氨水的浓度为0.001~0.1mol/l;所述炔基功能化共价三嗪聚合物与混合溶液的比例为(0.5~2)mg:1ml。
优选的,所述超声处理的温度为5℃~20℃;超声处理的超声功率为100~200w;超声处理的超声频率为20~60khz;超声处理的时间为20~50h。
本发明还提供了一种炔基功能化共价三嗪聚合物,包括式(ii)所示的重复单元;
其中,n为1~5的整数。
本发明还提供了上述炔基功能化共价三嗪聚合物在光催化制过氧化氢中的应用。
本发明提供了一种炔基功能化共价三嗪聚合物的制备方法,包括:s1)将式(i)所示的对称双氰基有机小分子在保护气氛中、三氟甲磺酸的催化下进行环三聚反应,得到炔基功能化共价三嗪聚合物;其中,n为1~5的整数。与现有技术相比,本发明制备的炔基功能化共价三嗪化合物在可见光范围内具有较好的吸收,且能带结构满足可见光照射下催化氧气还原和水氧化生成过氧化氢的要求;同时该炔基功能化共价三嗪化合物易剥离,可通过简单的液相超声剥离的方法,得到可以在水中分散性极好的二维纳米片层材料,有利于光生电子空穴的分离与传输,提高了光催化制过催化制过氧化氢的性能;再者该制备方法简单,合成条件温和,耗时短,且不需要任何金属催化剂,成本低廉。
附图说明
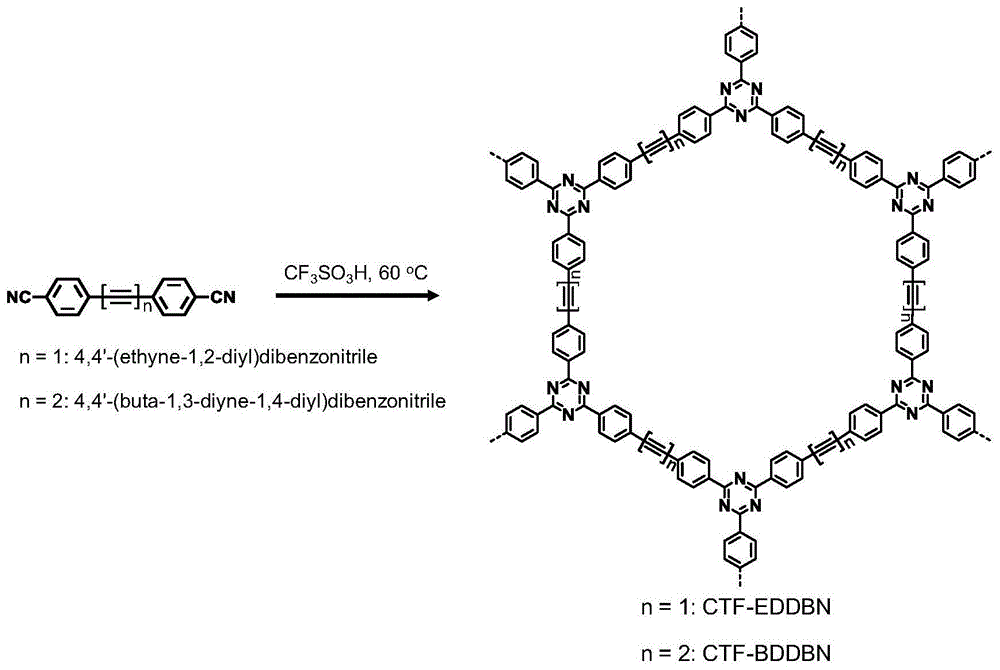
图1为本发明实施例1与实施例2制备炔基功能化共价三嗪聚合物的合成化学反应式;
图2a为本发明实施例1与实施例2中得到的炔基功能化共价三嗪聚合物的拉曼光谱图;
图2b为本发明实施例1与实施例2中得到的炔基功能化共价三嗪聚合物的红外光谱图;
图2c为本发明实施例1与实施例2中得到的炔基功能化共价三嗪聚合物的碳13固体核磁表征图;
图3a为本发明实施例1中得到的炔基功能化共价三嗪聚合物的二维纳米片的透射电子显微镜图;
图3b为本发明实施例2中得到的炔基功能化共价三嗪聚合物的二维纳米片的透射电子显微镜图;
图4a为本发明实施例1中得到的炔基功能化共价三嗪聚合物的二维纳米片的光学带隙图;
图4b为本发明实施例2中得到的炔基功能化共价三嗪聚合物的二维纳米片的光学带隙图;
图5为本发明实施例1与实施例2中得到的炔基功能化共价三嗪聚合物的二维纳米片在纯水氧饱和氛围下产过氧化氢性能图。
具体实施方式
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种炔基功能化共价三嗪聚合物,包括式(ii)所示的重复单元;
其中,n为1~5的整数,优选为1~3的整数,更优选为1或2。
在式(ii)中的波浪线代表炔基功能化共价三嗪聚合物中的重复单元或重复片段,其与本领域技术人员熟知的两边带波浪线的聚合物含义相同。
本发明还提供了一种上述炔基功能化共价三嗪聚合物的制备方法,包括:
一种炔基功能化共价三嗪聚合物的制备方法,其特征在于,包括:
s1)将式(i)所示的对称双氰基有机小分子在保护气氛中、三氟甲磺酸的催化下进行环三聚反应,得到炔基功能化共价三嗪聚合物;
其中,n为1~5的整数,优选为1~3的整数,更优选为1或2,即本发明中所述式(i)所示的对称双氰基有机小分子最优选如式(iii)或式(iv)所示。
在本发明中,上述步骤优选具体为:将式(i)所示的对称双氰基有机小分子与三氟甲磺酸在保护气氛中、低温条件下混合反应,然后转移至室温进行反应,最后进行热处理,得到炔基功能化共价三嗪聚合物。
将式(i)所示的对称双氰基有机小分子与所述三氟甲磺酸在保护气氛中、低温条件下混合反应;其中,式(i)所示的对称双氰基有机小分子与三氟甲磺酸的比例优选为(10~300)mg:1ml,更优选为(50~200)mg:1ml,再优选为(80~150)mg:1ml,再优选为(90~120)mg:1ml,最优选为(90~100)mg:1ml;所述保护气氛为本领域技术人员熟知的保护气氛即可,并无特殊的限制,本发明中优选为氩气;所述低温条件优选为0℃及以下,更优选为0℃~-10℃,再优选为-2℃~-8℃,再优选为-4℃~-6℃,最优选为-5℃;所述混合反应的时间优选为10~30min,更优选为15~25min,再优选为20min。
然后转移至室温进行反应;所述反应的时间优选为60~120min,更优选为80~100min,再优选为90min。
室温反应结束后,进行热处理;所述热处理的温度优选为50℃~80℃,更优选为55℃~70℃,再优选为60℃~65℃,最优选为60℃;所述热处理的时间优选为20~60min,更优选为30~50min,再优选为35~45min,最优选为40min。
热处理后,优选加入去离子水淬灭反应,更优选加入冷的去离子水淬灭反应。
淬灭反应后,优选用氨水与乙醇洗涤产物;所述氨水的浓度优选为0.1~1mol/l,更优选为0.3~0.8mol/l,再优选为0.4~0.6mol/l,最优选为0.5mol/l。
洗涤之后,优选用索氏提取法纯化产物,即将产物用提取溶剂进行纯化;通过提取溶剂进行索氏提取可将产物中的杂质抽提除去;所述索氏提取所用的提取溶剂优选依次为去离子水、乙醇、乙腈与丙酮;每种溶剂索提的时间优选为10~18h,更优选为10~14h,再优选为12h。
纯化之后,优选将产物进行干燥,得到炔基功能化共价三嗪聚合物;所述干燥的方法优选为真空干燥;所述真空干燥的温度优选为50℃~80℃,更优选为55℃~70℃,再优选为60℃~65℃,最优选为60℃;所述真空干燥的时间优选为20~30h,更优选为22~28h,再优选为24~26h,最优选为24h。
按照本发明,优选将所述炔基功能化共价三嗪聚合物通过液相超声进行剥离,更优选具体为:将所述炔基功能化共价三嗪聚合物分散于有机溶剂与氨水的混合溶液中,超声处理,离心后,得到炔基功能化共价三嗪聚合物的二维纳米片;所述有机溶剂优选为乙腈;所述氨水的浓度优选为0.001~0.1mol/l,更优选为0.005~0.05mol/l,再优选为0.01~0.02mol/l,最优选为0.01mol/l;所述有机溶剂与氨水的体积比优选为(2~10):1,更优选为(3~8):1,再优选为(4~6):1,再优选为(4~5):1,最优选为4:1;所述炔基功能化共价三嗪聚合物与混合溶液的比例优选为(0.5~2)mg:1ml,更优选为(0.5~1.5)mg:1ml,再优选为1mg:1ml;所述超声处理的超声功率优选为100~200w,更优选为140~180w,再优选为150~160w,最优选为150w;所述超声处理的超声频率优选为20~60khz,更优选为30~50khz,再优选为40khz;所述超声处理的温度优选为5℃~20℃,更优选为5℃~15℃,再优选为8℃~12℃,最优选为10℃;所述超声处理的时间优选为20~50h,更优选为30~45h,再优选为35~40h,最优选为36h;所述离心优选先进行低速离心,然后再进行高速离心;所述低速离心的转速优选为1000~5000rpm,更优选为2000~4000rpm,再优选为3000rpm;所述低速离心的时间优选为5~20min,更优选为5~15min,再优选为10min;通过低速离心可使未剥离的纳米片与聚集的纳米片沉积到底部;所述高速离心的转速优选为10000~20000rpm,更优选为12000~18000rpm,再优选为14000~16000rpm;所述高速离心的时间优选为5~20min,更优选为12min;通过高速离心可收集超薄的二维纳米片。
本发明制备的炔基功能化共价三嗪化合物在可见光范围内具有较好的吸收,且能带结构满足可见光照射下催化氧气还原和水氧化生成过氧化氢的要求;同时该炔基功能化共价三嗪化合物易剥离,可通过简单的液相超声剥离的方法,得到可以在水中分散性极好的二维纳米片层材料,有利于光生电子空穴的分离与传输,提高了光催化制过催化制过氧化氢的性能;再者该制备方法简单,合成条件温和,耗时短,且不需要任何金属催化剂,成本低廉。
本发明通过制备出不含任何金属的新型炔基功能化共价三嗪聚合物有机半导体催化剂实现了可见光催化水与氧气的反应制过氧化氢,是一种廉价、环保且可持续的光合成过氧化氢方式,因而该系列材料在未来太阳能转化领域展现出很大的潜力。
本发明还提供了一种上述炔基功能化共价三嗪聚合物在光催化制过氧化氢中的应用,更优选为可见光催化制过氧化氢。
在本发明中,所述光催化制过氧化氢的方法优选具体为:将炔基功能化共价三嗪聚合物的二维纳米片分散在水中,并持续鼓入氧气达到氧饱和,在波长大于420nm的可见光照射下,催化水和氧气反应制备过氧化氢。
本发明制备的新型炔基功能化共价三嗪聚合物材料在可见光范围具有较好的吸收,且能带结构满足可见光照射下催化氧气还原和水氧化生成过氧化氢的要求,实现了一种非常新颖的合成过氧化氢的方式。
为了进一步说明本发明,以下结合实施例对本发明提供的一种炔基功能化共价三嗪聚合物、其制备方法及应用进行详细描述。
以下实施例中所用的试剂均为市售。
实施例1
在-5℃,氩气保护气氛围下,将2.23毫升三氟甲磺酸缓慢滴加到含有200毫克1,2-双(4-氰基苯基)乙炔(如式iii所示记为eddbn)的反应瓶中,磁子搅拌二十分钟后,转移到室温25℃下,继续反应1.5小时,最后再进行60℃下热处理40分钟,反应停止,加入冷的去离子水淬灭反应,并用过量稀释的氨水(浓度0.5mol/l)和乙醇分别洗涤产物,接着用索氏提取的方法进行纯化,具体为将获得的产物用滤纸包裹好,放入抽体桶中,在250ml的抽提烧瓶内每次加入200ml的提取溶剂,每种溶剂各索提12h,且提取液不收集,作为废液处理,提取溶剂依次为去离子水,乙醇,乙腈和丙酮,索提结果后置于60℃真空干燥24小时,所获得的产物用研磨研磨成粉末,得到炔基功能化共价三嗪聚合物记为ctf-eddbn。
实施例2
在-5℃,氩气保护气氛围下,将2毫升1,4-双(4-氰基苯基)丁二炔缓慢滴加到含有200毫克1,2-双(4-氰基苯基)乙炔(如式iv所示,记为bddbn)的反应瓶中,磁子搅拌二十分钟后,转移到室温25℃下,继续反应1.5小时,最后再进行60℃下热处理40分钟,反应停止,加入冷的去离子水淬灭反应,并用过量稀释的氨水(浓度0.5m0l/l)和乙醇分别洗涤产物,接着用索氏提取的方法进行纯化,具体为将获得的产物用滤纸包裹好,放入抽体桶中,在250ml的抽提烧瓶内每次加入200ml的提取溶剂,每种溶剂各索提12h,且提取液不收集,作为废液处理,提取溶剂依次为去离子水,乙醇,乙腈和丙酮,索提结果后置于60℃真空干燥24小时,所获得的产物用研磨研磨成粉末,得到炔基功能化共价三嗪聚合物记为ctf-bddbn。
图1是实施例1与实施例2制备炔基功能化共价三嗪聚合物的合成化学反应式。
利用拉曼光谱对实施例1与实施例2中得到的炔基功能化共价三嗪聚合物进行分析,得到其拉曼光谱图如图2a所示。由图2a可以看到三氟甲磺酸催化后材料的氰基拉曼信号消失,而含有单炔和双炔结构的拉曼信号在合成反应后仍然保留。
利用红外光谱对实施例1与实施例2中得到的炔基功能化共价三嗪聚合物进行分析,得到其红外光谱图如图2b所示,可以从这两种材料的红外谱中看到三嗪结构的红外吸收峰,说明三嗪结构成功地通过实施例1、2形成,同时单炔双炔结构的红外吸收也能在图上看到,说明合成的三嗪材料结构保留完整。
利用核磁共振对实施例1与实施例2中得到的炔基功能化共价三嗪聚合物进行分析,得到其碳13固体核磁表征图如图2c所示。由图2c的结果说明了通过实施例1、2的确得到了理论的共价三嗪有机框架。
实施例3
将上述实施例1或实施例2中得到的研磨后的粉末100毫克,分散在80毫升乙腈和20毫升浓度为0.01摩尔/升的氨水混合溶液中,再将分散液置于超声机中进行液相超声剥离(超声机功率为150瓦,频率为40千赫兹),并且保持水温为10℃,超声36小时后,将分散液以3000rpm的转速离心十分钟,使未剥离的纳米片和聚集的纳米片沉积到底部,最后再将分散液以14000rpm转速离心收集超薄的二维纳米片。
利用透射电子显微镜对实施例3中得到的炔基功能化共价三嗪聚合物的二维纳米片进行分析,得到其透射电子显微镜图如图3所示,其中图3a为实施例1中得到的炔基功能化共价三嗪聚合物的二维纳米片;图3b为实施例2中得到的炔基功能化共价三嗪聚合物的二维纳米片。由图3可以看出每种材料剥离后都获得了约100纳米尺寸的二维片层形貌。
利用紫外可见近红外光漫反射光谱对实施例3中得到的炔基功能化共价三嗪聚合物的二维纳米片进行分析,得到其光学带隙图如图4所示,其中图4a为实施例1中得到的炔基功能化共价三嗪聚合物的二维纳米片;图4b为实施例2中得到的炔基功能化共价三嗪聚合物的二维纳米片。由图4a与图4b可说明获得三嗪系列的二维纳米片在可见光区域具有较好的吸收,ctf-eddbn与ctf-bddbn各自的光学带隙分别为2.78ev、2.43ev,说明其具备作为光催化剂使用的能力。
实施例4
将实施例3中剥离得到的二维纳米片材料30毫克加入到装有50毫升去离子水的石英瓶中,将分散液超声5分钟使材料在水中获得较好的分散性,然后持续通入氧气20分钟后,并用橡胶塞封住瓶口。光照之前先在黑暗中先搅拌30分钟使材料对氧气达到吸附平衡。光催化实验是用300瓦功率的氙灯作为光源,并配置一个420纳米的滤光片来获得可见光(λ>420纳米)。
每隔8小时,从光催化装置中吸取出3毫升的水作为检测样品,以14000rpm的转速离心样品来分离出光催化剂,之后过0.22微米的滤膜来进一步出去残留的光催化剂,将得到的样品与硫酸铈混合,待反应10min后,利用紫外吸收光谱检测硫酸铈在316纳米处的吸光度。
过氧化氢的检测是利用正4价的硫酸铈与催化生成的过氧化氢反应,正4价的离子被过氧化氢还原生成正三价的铈离子,再利用紫外光谱检测硫酸铈溶液316纳米处的吸收度变化,计算消耗的正4价离子的浓度,进而获得过氧化氢的浓度。
图5是实施例4中ctf-eddbn与ctf-bddbn二维纳米片各自的产过氧化氢性能图,由图5可看出24小时后分别产生了约40、70微摩尔的过氧化氢。
- 还没有人留言评论。精彩留言会获得点赞!