一种吡啶衍生物的制备方法及用途与流程
本发明属于药物领域,具体涉及一种吡啶衍生物的制备方法及用途。
背景技术:
常见的二氢吡啶衍生物类药物有氨氯地平、硝苯地平、尼卡地平、尼莫地平、尼群地平等三十余种,通常二氢吡啶衍生物类药物易氧化脱氢芳构化,生成吡啶衍生物,从而失去药用价值,因此各国药典中均对二氢吡啶衍生物类药物的氧化杂质吡啶衍生物有着严格的限度控制。
目前对于二氢吡啶衍生物类药物中的杂质吡啶衍生物的制备方法大多存在反应周期较长、产物组分较多,提纯处理较繁琐且产物收率低等缺点,因此确定一种吡啶衍生物的制备方法对有效控制二氢吡啶衍生物类药物具有重要意义。
技术实现要素:
本发明的目的在于提供一种吡啶衍生物的制备方法及用途,反应原料易得、操作简便、反应时间较短、生成产物单一且收率较高,对有效控制二氢吡啶衍生物类药物具有重要意义。
为实现上述发明目的,本发明采用的技术方案如下:
本发明一方面提供一种吡啶衍生物的制备方法,包括:
将二氢吡啶衍生物溶于反应溶剂中;
在所述反应溶剂中加入催化剂和氧化剂进行反应;
通过有机溶剂对所述反应溶剂进行萃取,得吡啶衍生物。
本发明提供的吡啶衍生物的制备方法,以二氢吡啶衍生物为原料,在催化剂和氧化剂的作用下,在反应溶剂中进行反应生成吡啶衍生物,该制备方法反应原料易得、操作简便、反应时间较短、生成产物单一且收率较高,对有效控制二氢吡啶衍生物类药物具有重要意义。
本发明另一方面提供了一种吡啶衍生物的用途,利用上述制备方法得到的吡啶衍生物纯度较高,可以用于在二氢吡啶衍生物的有关物质检查时作为标准对照品。
附图说明
图1为本发明实施例1制得的吡啶衍生物的核磁共振氢谱谱图;
图2为本发明实施例1制得的吡啶衍生物的质谱谱图。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
一方面,本发明实施例提供了一种吡啶衍生物的制备方法,包括:
步骤s10,将二氢吡啶衍生物溶于反应溶剂中;
步骤s20,在所述反应溶剂中加入催化剂和氧化剂进行反应;
步骤s30,通过有机溶剂对所述反应溶剂进行萃取,得吡啶衍生物。
本发明实施例提供的吡啶衍生物的制备方法,以二氢吡啶衍生物为原料,在催化剂和氧化剂的作用下,在反应溶剂中进行反应生成吡啶衍生物,该制备方法反应原料易得、操作简便、反应时间较短、生成产物单一且收率较高,对有效控制二氢吡啶衍生物类药物具有重要意义。
在一个实施例中,在步骤s10中,所述二氢吡啶衍生物包括如下式ⅰ所示化合物:
其中,取代基r1包括:-h或-ch3;取代基r2包括:-h、-ch3、-cooch3、-coo(ch2)2oc3h7、-coo(ch2)2och3、-cooch2ococ3h7、
上述取代基r1、取代基r2、取代基r3、取代基r4、取代基r5、取代基r6和取代基r7可以任意组合,例如当r1为-ch3,r2为-cooch3,r3为
此时,吡啶衍生物的结构可如下式ⅱ所示:
在一个实施例中,在步骤s10中,所述二氢吡啶衍生物包括如下式ⅰ所示化合物的盐:
其中,取代基r1、取代基r2、取代基r3、取代基r4、取代基r5中至少一个取代基可以与盐酸、硫酸或苯磺酸结合制成相应的盐酸盐、硫酸盐或苯磺酸盐,例如盐酸尼卡地平和苯磺酸氨氯地平等。当二氢吡啶衍生物为盐酸盐、硫酸盐或苯磺酸盐时,所述通过有机溶剂对所述反应溶剂进行萃取,得吡啶衍生物包括:通过有机溶剂对所述反应溶剂进行萃取,成盐,得吡啶衍生物。具体地,当原料为盐酸尼卡地平时,通过有机溶剂对反应溶剂进行萃取,在萃取结束后,有机溶剂分别通过水和盐水洗涤,干燥剂干燥,去除干燥剂,对有机溶剂减压浓缩,所得残余物中加入甲基叔丁基醚,并在室温下缓慢通入盐酸气成盐搅拌过夜,得吡啶衍生物。
进一步地,在步骤s10中,所述反应溶剂包括甲醇、乙醇、异丙醇、叔丁醇或四氢呋喃中的一种溶剂或多种的混合溶剂,二氢吡啶衍生物在上述反应溶剂中有较好的溶解性,可以保证反应的正常进行。优选地,所述反应溶剂为甲醇或乙醇。
进一步地,在步骤s20中,所述催化剂包括钨酸钠、五氧化二钒、二氧化锰、氧化铬、氧化锰、氧化铁、氧化钴、氧化镍、氧化铜、氧化亚铜或氧化锌中的一种或多种,以上催化剂均可以使得二氢吡啶衍生物在反应溶剂中氧化剂的作用下生成吡啶衍生物。优选地,所述催化剂包括五氧化二钒、二氧化锰或氧化铁,当催化剂为五氧化二钒、二氧化锰或氧化铁时可以使得吡啶衍生物的收率更好。
进一步地,在步骤s20中,所述氧化剂包括双氧水、次氯酸钠或次氯酸钙,以上氧化剂均可以将二氢吡啶衍生物在催化剂作用下制成吡啶衍生物。优选地,所述氧化剂为双氧水,使用双氧水作为氧化剂得到的吡啶衍生物收率较高。
进一步地,在步骤s20中,所述氧化剂与所述二氢吡啶衍生物的摩尔比为1~10,例如,氧化剂与二氢吡啶衍生物的摩尔比可以为1、6、7、8、10等。通常一定浓度的氧化剂才可以提供所需要的反应速率,当氧化剂与二氢吡啶衍生物的摩尔比过小时,氧化剂浓度过低,反应速率过慢,且反应结束后,反应原料二氢吡啶衍生物可能会有剩余;当氧化剂与二氢吡啶衍生物的摩尔比过大时,大量氧化剂的存在可能生成副产物,即生成吡啶衍生物后继续进行反应,从而影响吡啶衍生物的纯度和收率,本实施例中,控制氧化剂与二氢吡啶衍生物的摩尔比为1~10,可以保证反应时间较短、且避免副产物的生成。优选地,所述氧化剂与所述二氢吡啶衍生物的摩尔比为1.5~5,例如,氧化剂与二氢吡啶衍生物的摩尔比为1.5、2、2.5、3、4、5等,在此摩尔比下吡啶衍生物的收率更高。
进一步地,在步骤s20中,控制反应温度为-25℃~80℃,例如,反应温度可以为-25℃、-10℃、5℃、15℃、60℃或80℃等,通常反应温度会直接影响反应速率,当反应温度过低时,反应速度过慢,需要较长的时间才能完成反应;当反应的温度过高时,可能会生成副产物影响吡啶衍生物的纯度,本实施例中,控制反应温度为-25℃~80℃,可以保证反应较快的进行,同时避免副产物的生成。优选地,控制反应温度为20℃~50℃,例如,反应温度可以为20℃、25℃、30℃、35℃、40℃或50℃等,在此温度范围下,可以更快的结束反应过程,且吡啶衍生物的纯度较高。
进一步地,在步骤s20后、步骤s30前,还包括:利用薄层色谱法对反应进程进检测,可以掌握反应结束时间,使得原料反应完全,如果不对反应进程进行监测,可能在原料未完全反应时结束反应进程,降低吡啶衍生物的收率并影响吡啶衍生物的纯度;如果通过等待较长时间使得反应完全,可能会生成副产物,即生成的吡啶衍生物会继续氧化,生成吡啶氮氧化物,从而影响吡啶衍生物的收率和质量,本实施例,利用薄层色谱法对反应进程进检测,可以适当的缩短反应时间,且避免副产物的生成。
进一步地,在步骤s20后、在步骤s30前,还包括向反应溶剂中加入淬灭剂,其中,淬灭剂包括亚硫酸钠、硫代硫酸钠或亚硫酸氢钠。在氧化反应结束后,加入淬灭剂对氧化剂进行淬灭,可以保证反应进程的安全,在加入淬灭剂的过程中,至少加入与氧化剂等摩尔量的淬灭剂,大于氧化剂摩尔量的淬灭剂的淬灭效果更好,其中淬灭剂可以为亚硫酸钠、硫代硫酸钠或亚硫酸氢钠等还原性溶液。具体地,在加入淬灭剂后,在室温条件下至少搅拌5个小时,以保证氧化剂淬灭完全,其中5个小时为经验值;或者,可以对溶液进行残余氧检测,如利用淀粉碘化钾试纸检测淬灭程度,确定氧化剂被完全淬灭后,进行后续处理。
进一步地,在步骤s30中,有机溶剂可以为二氯甲烷,具体地,可以利用150ml二氯甲烷分多次对反应溶剂进行萃取,萃取后,合并有机相,然后分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物经过重结晶或成盐得吡啶衍生物。
本发明实施例的吡啶衍生物的制备方法的一种优选步骤为:
步骤s1,将二氢吡啶衍生物溶于反应溶剂中。
步骤s2,准备与二氢吡啶衍生物的摩尔比为1.5~5的双氧水,将催化剂和准备好的双氧水加入反应溶剂中进行反应,控制反应温度为20℃~50℃,并利用薄层色谱法对反应进程进检测。
步骤s3,向所述反应溶剂中加入淬灭剂,搅拌至少5小时,过滤、滤液进行浓缩后,利用二氯甲烷分多次对滤液进行萃取,萃取后,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物经过重结晶或成盐得吡啶衍生物。
另一方面,本发明实施例提供了一种吡啶衍生物的用途,利用上述制备方法得到的吡啶衍生物纯度较高,用于在二氢吡啶衍生物有关物质检查时作为标准对照品。
本发明先后进行过多次试验,现举一部分试验结果作为参考,对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
实施例1
步骤s1,将10g硝苯地平(28.9mmol)溶于150ml甲醇中。
步骤s2,加入0.1g五氧化二钒和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度20℃搅拌反应,反应1小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约43℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时,过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入200ml正庚烷打浆过夜,过滤并烘干得淡黄色固体9.2g,收率92.56%。
对所得淡黄色固体产物进行鉴定:
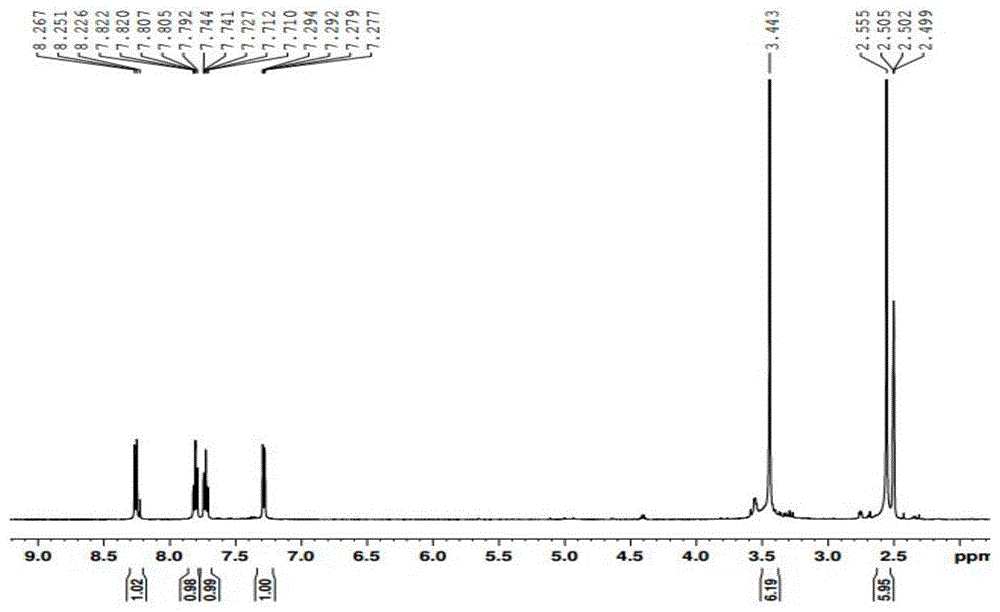
淡黄色固体产物的核磁氢谱谱图如图1所示,
1h-nmr(500mhz,dmso-d6)δ8.251~8.267(d,3h),7.792~7.822(m,1h),7.710~7.744(m,1h),7.277~7.294(dd,1h),3.443(s,6h),2.555(s,6h)。
淡黄色固体产物的质谱谱图如图2所示,得到白色固体产物的lc-msm/z:345.11[m+h]+、711.2[2m+na]+。
因此可确定上述淡黄色固体为硝苯地平氧化脱氢芳构化的吡啶衍生物。
淡黄色固体产物的纯度检测:
以十八烷基硅烷键合硅胶为填充剂,甲醇-水(60:40)为流动相,235nm为检测波长,记录色谱图至硝苯地平峰保留时间的2倍,具体参见《中国药典》2015版第二部高效液相色谱法,测定淡黄色固体的纯度为98.85%。
实施例2
步骤s1,将20g盐酸尼卡地平(38.76mmol)溶于200ml甲醇中。
步骤s2,加入0.1g五氧化二钒和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度20℃搅拌反应,反应约0.5小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约45℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入300ml甲基叔丁基醚,室温下缓慢通入盐酸气成盐并搅拌过夜,过滤并烘干得白色固体吡啶衍生物18.6g,收率93.37%。
利用《中国药典》2015版第二部高效液相色谱法对白色固体的纯度进行检测,测定白色固体的纯度为99.45%。
实施例3
步骤s1,将20g苯磺酸氨氯地平(35.27mmol)溶于200ml甲醇中。
步骤s2,加入0.2g五氧化二钒和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度20℃搅拌反应,反应约4.5小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约22℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入300ml甲基叔丁基醚,室温下缓慢通入盐酸气成盐并搅拌过夜,过滤并烘干得白色固体吡啶衍生物13.2g,收率84.4%。
利用《中国药典》2015版第二部高效液相色谱法对白色固体的纯度进行检测,测定白色固体的纯度为99.65%。
实施例4
步骤s1,将10g尼莫地平(23.9mmol)溶于150ml甲醇中。
步骤s2,加入0.1g五氧化二钒和12.0g质量分数为30%的双氧水溶液(106mmol的过氧化氢),控制反应温度25℃搅拌反应,反应约1小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约27℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入250ml甲基叔丁基醚,室温下缓慢通入盐酸气成盐并搅拌过夜,过滤并烘干得白色固体吡啶衍生物9.2g,收率84.4%。
利用《中国药典》2015版第二部高效液相色谱法对白色固体的纯度进行检测,测定白色固体的纯度为99.65%。
实施例5
步骤s1,将20g盐酸尼卡地平(38.76mmol)溶于150ml甲醇中。
步骤s2,加入0.5g三氧化二铁和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度20℃搅拌反应,反应约4.5小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约22℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入300ml甲基叔丁基醚,室温下缓慢通入盐酸气成盐并搅拌过夜,过滤并烘干得白色固体吡啶衍生物16.8g,收率83.34%。
利用《中国药典》2015版第二部高效液相色谱法对白色固体的纯度进行检测,测定白色固体的纯度为98.68%。
实施例6
步骤s1,将10g硝苯地平(28.9mmol)溶于150ml甲醇中。
步骤s2,加入0.5g钨酸钠和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度20℃搅拌反应,反应约4.5小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约43℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入200ml正庚烷打浆过夜,过滤并烘干得淡黄色固体吡啶衍生物9.2g,收率92.56%。
利用《中国药典》2015版第二部高效液相色谱法对淡黄色固体的纯度进行检测,测定淡黄色固体的纯度为98.45%。
实施例7
步骤s1,将10g硝苯地平(28.9mmol)溶于150ml甲醇中。
步骤s2,加入0.2g五氧化二钒和50g次氯酸钠溶液(约79mmol有效氯),控制反应温度20℃搅拌反应,反应约2.5小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约21℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入200ml正庚烷打浆过夜,过滤并烘干得淡黄色固体吡啶衍生物8.8g,收率88.54%。
利用《中国药典》2015版第二部高效液相色谱法对淡黄色固体的纯度进行检测,测定淡黄色固体的纯度为97.85%。
实施例8
步骤s1,将10g硝苯地平(28.9mmol)溶于150ml甲醇中。
步骤s2,加入0.5g钨酸钠和次氯酸钙的水溶液(约114mmol有效氯),控制反应温度20℃搅拌反应,反应4.5小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约43℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入200ml正庚烷打浆过夜,过滤并烘干得淡黄色固体吡啶衍生物7.6g,收率77.56%。
利用《中国药典》2015版第二部高效液相色谱法对淡黄色固体的纯度进行检测,测定淡黄色固体的纯度为98.32%。
实施例9
步骤s1,将20g盐酸尼卡地平(38.76mmol)溶于200ml乙醇中。
步骤s2,加入0.5g二氧化锰和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度20℃搅拌反应,反应约0.7小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约43℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入300ml甲基叔丁基醚,室温下缓慢通入盐酸气成盐并搅拌过夜,过滤并烘干得白色固体吡啶衍生物18.4g,收率92.37%。
利用《中国药典》2015版第二部高效液相色谱法对白色固体的纯度进行检测,测定白色固体的纯度为99.56%。
实施例10
步骤s1,将20g盐酸尼卡地平(38.76mmol)溶于200ml四氢呋喃中。
步骤s2,加入0.1g氧化铁和6.6g质量分数为30%的双氧水溶液(58.14mmol的过氧化氢),控制反应温度50℃搅拌反应,反应约0.5小时利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成;此时反应溶剂温度约53℃。
步骤s3,向反应溶剂中加入50ml亚硫酸氢钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入300ml甲基叔丁基醚,室温下缓慢通入盐酸气成盐并搅拌过夜,过滤并烘干得白色固体吡啶衍生物18.2g,收率91.36%。
利用《中国药典》2015版第二部高效液相色谱法对白色固体的纯度进行检测,测定白色固体的纯度为99.12%。
实施例11
步骤s1,将10g硝苯地平(28.9mmol)溶于150ml异丙醇中。
步骤s2,加入0.1g五氧化二钒和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度80℃搅拌反应,反应5分钟利用薄层色谱法对反应进程进检测,原料消失,有小极性新物质生成(同时观察到有微量大极性杂质点生成);此时反应溶剂温度约82℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入200ml正庚烷打浆过夜,过滤并烘干得淡黄色固体吡啶衍生物7.6g,收率76.5%。
利用《中国药典》2015版第二部高效液相色谱法对淡黄色固体的纯度进行检测,测定淡黄色固体的纯度为95.05%。
实施例12
步骤s1,将10g硝苯地平(28.9mmol)溶于150ml叔丁醇中。
步骤s2,加入0.1g五氧化二钒和3.3g质量分数为30%的双氧水溶液(29mmol的过氧化氢),控制反应温度70℃搅拌反应,反应4小时利用薄层色谱法对反应进程进检测,原料有微量残留,有小极性新物质生成;此时反应溶剂温度约71℃。
步骤s3,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入200ml正庚烷打浆过夜,过滤并烘干得淡黄色固体吡啶衍生物7.8g,收率78.5%。
利用《中国药典》2015版第二部高效液相色谱法对淡黄色固体的纯度进行检测,测定淡黄色固体的纯度为95.65%。
实施例13
步骤s1,将20g盐酸尼卡地平(38.76mmol)溶于200ml乙醇中。
步骤s2,加入0.1g五氧化二钒和43.8g质量分数为30%的双氧水溶液(387.6mmol的过氧化氢),控制反应温度-20℃搅拌反应,反应约15小时利用薄层色谱法对反应进程进检测,原料基本消失,有小极性新物质生成;此时反应溶剂温度约-18℃。
步骤s3,升温到室温,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入300ml甲基叔丁基醚,室温下缓慢通入盐酸气成盐并搅拌过夜,过滤并烘干得白色固体吡啶衍生物17.8g,收率89.36%。
利用《中国药典》2015版第二部高效液相色谱法对白色固体的纯度进行检测,测定白色固体的纯度为96.39%。
实施例14
步骤s1,将10g硝苯地平(28.9mmol)溶于150ml甲醇中。
步骤s2,加入0.1g五氧化二钒和16.4g质量分数为30%的双氧水溶液(145mmol的过氧化氢),控制反应温度-25℃搅拌反应,反应约10小时利用薄层色谱法对反应进程进检测,原料大量剩余,有小极性新物质生成;继续控制此温度下反应56h,原料消失。
步骤s3,升温至室温,向反应溶剂中加入50ml饱和硫代硫酸钠水溶液,并维持在室温条件下至少搅拌5小时;混合溶液过滤,滤液进行浓缩后,利用150ml二氯甲烷分两次对滤液进行萃取,合并有机相,分别利用水和盐水对有机相进行洗涤,并利用干燥剂进行干燥,去除干燥剂,对有机相进行减压浓缩,所得残余物加入200ml正庚烷打浆过夜,过滤并烘干得淡黄色固体吡啶衍生物9.2g,收率89.54%。
利用《中国药典》2015版第二部高效液相色谱法对淡黄色固体的纯度进行检测,测定淡黄色固体的纯度为97.35%。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!