一种铁皮石斛精制多糖及其制备方法与流程
本发明涉及药物提取领域,特别是一种铁皮石斛精制多糖及其制备方法。
背景技术:
铁皮石斛(dendrobiumofficinale)是一种兰科植物,又名黑节草,为多年生附生型草本植物,长于海拔达1600米的山地半阴湿的岩石上,在自然野生状态下萌发率低,且必需与真菌共生才能萌发,再加上其生长缓慢,野生石斛已经很少,已被列为我国重点保护的珍惜濒危药用植物。主要分布我国的广西、浙江、云南、安徽等地。《中华人民共和国药典》中记载铁皮石斛具有益胃生津,滋阴清热的功能。多糖、氨基酸、石斛碱是铁皮石斛的主要有效成分,现代研究表明铁皮石斛有增强免疫、抗疲劳、抗氧化、促消化、降血糖、降血压、抗肝损伤、抗肿瘤等方面药理作用。因而具有“药中黄金”、“救命仙草”的美誉。
铁皮石斛与其他石斛相比,多糖含量较高,药理活性更强,在免疫调节、抗氧化、抗衰老、抗癌、抗肝损伤等方而具有良好的应用前景。但目前一些方而研究水平相对简单,主要表现在研究对象多为粗多糖,杂质较多,难以在分子水平上阐述其药用机理,并且多糖分子结构难以准确得出。铁皮石斛多糖与其它石斛多糖的专属性鉴别研究较少,质量难以控制。因此,铁皮石斛多糖的纯化工艺以及专属性鉴别研究应该是今后的研究方向,以确保铁皮石斛多糖产品的质量。
技术实现要素:
为了克服现有技术存在的不足,本发明提供一种铁皮石斛精制多糖及其制备方法。
本发明所述铁皮石斛,均为兰科植物dendrobiumofficinale的茎。
本发明的目的可以通过以下技术方案来实现:
一种如式ⅰ所示的铁皮石斛精制多糖
式ⅰ。
本发明还提供上述式ⅰ所示的铁皮石斛精制多糖的制备方法,包括如下步骤:
(1)将铁皮石斛茎,加水,80℃提取2h,提取两次,浓缩,加入95%乙醇,4℃下醇沉48h,离心分离沉淀,冷冻干燥得到铁皮石斛多糖粗品;
(2)将步骤(1)所得的铁皮石斛多糖粗品溶于去离子水,加入氯化717阴离子交换树脂进行脱色,脱色后溶液离心取上清液,加入氯仿、正丁醇充分摇匀并震荡,离心除去出现的沉淀。重复多次加入氯仿、正丁醇的操作直至离心后无沉淀产生;冷冻干燥得到除蛋白样品;
(3)取步骤(2)所得的除蛋白样品溶于去离子水,0.45μm滤膜过滤除杂,上deae-52纤维素层析柱,用280ml去离子水进行洗脱,流速1ml/min,收集洗脱液,用3500da透析袋进行透析1天,浓缩,冷冻干燥,得到初步纯化多糖;
(4)取步骤(3)所得的初步纯化多糖溶于去离子水,0.45μm滤膜过滤除杂,上sephadexg-100凝胶层析柱,用100ml去离子水洗脱,流速0.5ml/min,弃去先流出的50ml,收集后流出的50ml,用3500da透析袋进行透析1天,浓缩,冷冻干燥,即得铁皮石斛精制多糖。
与现有技术比较,本发明的方法具有以下优点:
本发明的铁皮石斛精制多糖具有良好的抗氧化活性,尤其清除羟自由基能力良好。
附图说明
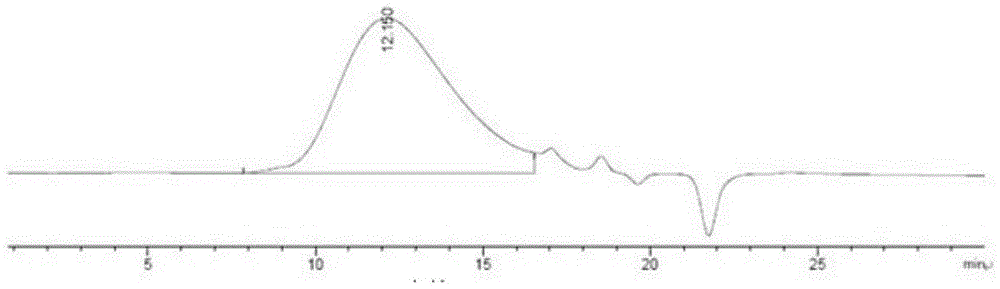
图1为dop的hplc-gpc图。
图2为dop衍生化后hplc图。
图3为dop红外光谱图。
图4为dop的1h-nmr谱。
图5为dop的13c-nmr谱。
图6为dop甲基化tic图。
图7为dop结构图。
图8为多糖清除abts自由基能力。
图9为多糖清除羟基自由基能力。
图10多糖总还原能力
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例1铁皮石斛精制多糖的制备
1.1主要材料
铁皮石斛,采自广西;抗坏血酸(vc)、2,2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(abts)等,均为国产分析纯;氯化717阴离子交换树脂,索莱宝科技有限公司;deae-52纤维素,大连美仑生物技术有限公司;sephadexg-100、3500da透析袋,上海源叶生物科技有限公司;d-(+)-葡萄糖、d-(+)-甘露糖、d-(+)-半乳糖(ar),阿拉丁试剂有限公司;l-鼠李糖,大连美仑生物技术有限公司;d-半乳糖醛酸,中国食品药品检定研究院。
1.2主要仪器旋转蒸发仪re-3000,上海亚荣生化有限公司;超声波清洗机,洁盟清洗设备有限公司;集热式恒温加热磁力搅拌器df-101s,巩义市予华仪器有限责任公司;离心机,上海安亭科学仪器厂;thermomultiskango酶标仪,美国赛默飞公司;真空冷冻干燥机lgj-10,北京松源华兴科技发展有限公司;高效液相色谱lc-20a,日本岛津仪器公司;hh数显恒温油浴锅,常州国宇仪器制造有限公司。
1.3方法
(1)将新鲜的铁皮石斛茎干燥粉末1g,加入30ml蒸馏水,80℃提取2h,提取两次,旋转蒸发仪浓缩至20ml,加入80ml95%乙醇,4℃下醇沉48h,离心分离沉淀,真空冷冻干燥得到铁皮石斛多糖粗品0.3156g;
(2)将步骤(1)所得的铁皮石斛多糖粗品溶于10ml去离子水,加入氯化717阴离子交换树脂进行脱色,脱色后溶液离心取上清液,加入氯仿2ml、正丁醇0.5ml,充分摇匀,震荡,离心除去出现的沉淀。重复多次加入氯仿、正丁醇的操作直至离心后无沉淀产生。冷冻干燥得到除蛋白样品。
(3)取0.3g步骤(2)所得的样品溶于去离子水,0.45μm滤膜过滤除杂,上deae-52纤维素层析柱,用280ml去离子水进行洗脱,流速1ml/min,收集洗脱液,用3500da透析袋进行透析1天,浓缩,冷冻干燥,得到初步纯化多糖0.052g。
(4)取步骤(3)所得的初步纯化多糖0.050g铁皮石斛初步纯化多糖于2ml的去离子水,0.45μm滤膜过滤除杂,上sephadexg-100凝胶层析柱,用100ml去离子水洗脱,流速0.5ml/min,弃去先流出的50ml,收集后流出的50ml,用3500da透析袋进行透析1天,浓缩,冷冻干燥,即得铁皮石斛精制多糖0.024g,命名为dop。
实施例2分子量分析
分别称取不同分子量的葡聚糖标准品,加入0.002mol/l磷酸二氢钠(含0.05%nan3),将其配置成浓度约3mg/ml的对照品溶液,之后逐一进行gpc检测,根据标准糖mw的保留时间绘制标准曲线,统计线性关系。称取一定量的多糖样品,加入0.002mol/l磷酸二氢钠(含0.05%nan3),将其配制成浓度约为1mg/ml的溶液,进样检测。
根据标准糖重均分子量(mw)的保留时间得到标准曲线,y=-0.4394x+10.788,r2=0.9982。从图1中可以得出,dop的保留时间为12.150min,经以标准曲线计算,得到样品主要成分峰的mw为2.76x10^5。
实施例3组成分析
采用uv-vis检测器,色谱柱为c18柱(5μm,4.6mm×250mm),柱温30℃,流动相为磷酸盐缓冲液和乙腈以82:18的比例进行等度洗脱,进样体积为10μl,流速为1ml/min,检测波长为250nm。
如图2所示,高效液相分离得到两个峰,与标准单糖的液相色谱图进行比较,得出dop由甘露糖(man)和葡萄糖(glc)组成,计算其峰面积比后得到其摩尔比为14.72:1。
实施例4红外光谱分析
干燥的精制多糖样品1mg与50mg干燥的kbr晶体混匀研磨后压片,采用傅里叶变换红外光谱仪扫描分析(扫描范围400-4000cm-1)。
dop的红外光谱如图3,3379cm-1和1061.08cm-1吸收峰是分别由糖分子内或分子间氢键的羟基伸缩振动和变形振动引起,926.96cm-1为吡喃糖环结构,880.72cm-1和839.63cm-1表明该多糖含有α-d-甘露糖,1750-1700cm-1之间无吸收,表明不含糖醛酸,为中性糖,这与hplc单糖检测结果相符。
实施例5核磁分析
取精制多糖10mg溶于0.5mld2o中,离心后取上清于核磁管中,内标为tms,进行1h-nmr和13c-nmr分析。
1h-nmr多用于研究多糖糖苷键的构型特点,α型糖苷键的质子信号通常集中在大于5的位置,而β型糖苷键的化学位移一般低于5。多糖dop的核磁谱图如图4和图5,在1h-nmr谱中,异头区δ4.4-δ5.8范围内,有2个信号分别为δ5.32和δ5.12,说明这些残基是α构型的糖苷键,并有两种糖,这与红外光谱分析的结果是一致的。4.70处的信号为d2o所致;此外,dop的主要异头碳质子信号主要集中于3.1-4.55处,说明dop主要由α构型吡喃糖苷键连接,与ir中相吻合。由13c-nmr可以辨认异头碳类型,δc90-110范围内是异头碳的共振范围,其中小于δc100的为α型异头碳,有2个主要信号,分别为δc95.88、δc92.05,说明dop含有α构型的单糖组成,归属于α-d-man的异头碳,与ir结论一致。葡萄糖和甘露糖为六碳醛糖,δc62.31为ch2oh的化学位移。
实施例6单糖连接方式分析
取干燥的2mgdop样品置于干净的梨型瓶中,溶于1mldmso中,通氮气,超声片刻助溶,静置30min。加入0.6mlnaoh-dmso悬液(取0.5g细粉状naoh溶于1ml超纯水中,取0.2mlnaoh与0.2ml甲醇混均,然后用6mldmso稀释,剧烈振荡数分钟,然后超声5min,低速离心5min,收集naoh沉淀。重复三次,最后沉淀悬于4mldmso中即得naoh-dmso悬液。),通氮气并用保鲜膜封口,保持无氧状态,密闭混合超声30min。静置1h后,在通风、避光冰浴中用注射器缓慢滴加碘甲烷1ml,边注射边震荡,每次滴加持续时间约1min,之后室温避光超声20min。加入3ml水振荡终止反应,加入3ml氯仿,充分振荡,离心取下层有机相,并水洗5次。向氯仿相加入少量无水硫酸钠,充分混合,以除去残留的水分,将氯仿用一次性注射器吸取过0.45有机滤膜,收集滤液。残留物加入少量氯仿进行洗涤,洗液也经滤膜过滤。合并滤液置干净的梨型瓶中,40摄氏度下减压蒸发至干。
加入3ml2mol/l的tfa,玻璃塞塞紧,封口处缠绕保鲜膜密封加固,剧烈震荡。120℃下水解4h。冷却至室温,加入0.5ml甲醇于40℃下减压旋干,反复4次(反复时每次加入甲醇的量为2ml),以彻底去除tfa。加入1ml新配的10mg/mlnabh4,室温反应2h,期间振荡数次。逐滴加入4mol/l乙酸中和,ph试纸检测。加入1ml甲醇,减压蒸干,重复2次。加入1ml新配制的吡啶/乙酸酐(1:1),120℃反应30min。冷却至室温,减压蒸干。加入1ml甲醇,减压蒸干。加入2ml二氯甲烷,旋涡振荡,离心,取上清液,进行gc/ms分析。起始温度80℃,保持0.5min,以3℃/min升温至250℃,保持10min。载气为氦气,ei源,离子化电势70ev,进样量1μl,分流比1:10。
dop经甲基化、水解及乙酰化后,gc-ms得到两个主要峰,如图6中峰1和峰2,峰1为1,3,5,6-四-o-乙酰基-2,4-二-o-甲基-d-甘露糖醇,峰2为1,4,5-三-o-乙酰基-2,3,6-三-o-甲基-d-葡萄糖醇,根据检索质谱图和裂解碎片,两种糖的连接方式分别为→3,6)man-(1→、→4)glc-(1→。结合hplc单糖结果可以得出,dop主链由→3,6)man-(1→组成,侧链由末端连接的葡萄糖组成。由此推断多糖结构如图7所示。
实施例7清除abts自由基实验
配制浓度为0.1mg/ml、0.5mg/ml、1mg/ml、2mg/ml、3mg/ml、4mg/ml、5mg/ml的多糖溶液,取7mmol/l的abts与1.4mmol/l的过硫酸钾溶液等体积混合均匀,避光放置16h,临用前用蒸馏水稀释至吸光度为0.7±0.02,得abts工作液。取工作液150μl、样品溶液50μl,置于96孔板孔中,避光反应6min后于734nm波长处测定吸光度,记为a样品;以溶剂代替样品,同法测定吸光度,记为a空白;以150μl去离子水代替abts,同法测得各样品对照组的吸光度。以vc为阳性对照品,测定各浓度清除率。计算ec50。
清除率(%)=[(a空白-a样品+a对照)/a空白]×100
由图8可知,粗多糖和精制多糖dop浓度在0-5mg/ml浓度范围内,随着多糖浓度的增加,清除率也随之提高,呈明显剂量关系。粗多糖在5mg/ml时清除率达到53.06%,此时清除率曲线已趋于平缓;dop浓度为5mg/ml时,清除率34.36%。经过曲线拟合计算,粗多糖的ec50值是1.856mg/ml,精制多糖是4.500mg/ml。
实施例8清除羟自由基实验
分别量取200μl浓度为0.1mg/ml、0.5mg/ml、1mg/ml、2mg/ml、3mg/ml、4mg/ml、5mg/ml的多糖溶液,然后依次加入200μl2mmol/lfes04溶液,200μl2mmol/lh2o2溶液,混匀,室温反应10min,然后加入200μl2mmol/l水杨酸溶液,混匀,37℃恒温反应30min。空白组用蒸馏水代替铁皮石斛多糖溶液,对照组用蒸馏水代替水杨酸溶液。510nm处测量吸光度值,以vc为阳性对照,计算各组清除率,方法同上。计算ec50。
从图9中可以看出,在0-5mg/ml浓度范围内,粗多糖清除率明显高于精多糖。当浓度高于4mg/ml时,精多糖s和纯化多糖清除率曲线趋于平缓,5mg/ml时,粗多糖和纯化多糖最大清除率分别为84.61%和60.45%。阳性对照vc浓度在1mg/ml已经达到最大清除率。多糖对abts和羟自由基清除效果良好,经过计算,粗多糖和精制多糖ec50分别为1.626mg/ml和2.057mg/ml。
实施例9测定总还原能力
配制浓度为0.1mg/ml、0.5mg/ml、1mg/ml、2mg/ml、3mg/ml、4mg/ml、5mg/ml的多糖溶液,在2ml离心管中加入500μl0.2mol/l磷酸盐缓冲溶液(ph=6.6),200μl样品溶液,500μl1%铁氰化钾,立刻混匀后于50℃反应20min,加入500μl10%三氯乙酸,混匀静置反应10min,取出500μl,加入500μlh2o,100μl0.1%三氯化铁溶液,静置10min,于700nm波长处测定吸光度,用于表示还原能力的强度。
如图10所示,浓度为5mg/ml时,粗多糖和精制多糖吸光度值分别为0.89和0.58。
虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!