双靶点识别可调控工程化免疫细胞的制备方法与流程
本发明涉及采用双靶点识别分子提高嵌合抗原受体(car)-免疫细胞用于提高肿瘤治疗的安全性和有效性。
背景技术:
癌症治疗一直是临床医疗领域尚未攻克的难题之一,随着现代医学的发展,癌症的免疫治疗逐渐取得了一定的突破,并成为癌症治疗领域的焦点。2011年12月一篇名为《cancerimmunotherapycomesofage》的评论文章同时刊登在nature和临床肿瘤最顶级杂志journalofclinicaloncology上,文章评论免疫疗法是现有科技中唯一有可能彻底清除癌细胞的方法,该方法弥补了传统疗法的弊端,被认为是二十一世纪肿瘤综合治疗模式中最活跃、最有发展前途的一种治疗手段(1)。目前,免疫治疗手段包括抗体类药物、疫苗类药物、免疫检查点抑制剂、细胞因子以及过继性细胞疗法等,而过继性细胞治疗策略在对免疫细胞进行工程化改造后,无论在肿瘤治疗的靶向特异性和功能持久性等方面都得到了显著的提高,而2017年的两款嵌合抗原受体(car)-t细胞产品的获批,印证了工程化免疫细胞疗法也从治疗的一种前景概念发展成为现实的解决方案。
嵌合抗原受体修饰t细胞(chimericantigenreceptor-tcell,car-t)治疗肿瘤的方法是近年来发展最快的一种肿瘤免疫新疗法。car的修饰可使t细胞在获得靶向杀伤能力的同时获得更强的增殖及抗凋亡的能力,其肿瘤杀伤作用的发挥也不会受到主要组织相容性复合体(mhc)的限制。1989年,eshhar(2)及其团队第一次提出了“嵌合受体”的概念,他们将t细胞受体(tcr)的可变区用单链抗体(scfv)替代,与t细胞内信号传递结构域cd3ζ或是fcrγ进行组合,由此得到了具有抗原靶向性的t细胞,经改造后的t细胞不再具有mhc的限制性,上述嵌合受体的形式已经基本形成了第一代car-t细胞的雏形。目前,第一代car-t细胞技术已在淋巴瘤、鼻咽癌、卵巢癌及肾癌中进行了初步的临床试验,由于其在体内的存活时间并不理想,从而影响了最终的临床效果。为了保证car-t细胞能够完全活化,第二代及第三代car-t细胞引入了一个或两个共刺激信号的细胞内信号传递结构域(如cd28,4-1bb或者ox40),从而赋予了car-t细胞更强的增殖及抗凋亡能力,其细胞因子的分泌水平及细胞毒性同时也有所增强。然而由于肿瘤的混合异质性和免疫抑制微环境的存在,car-t细胞无法对复杂实体肿瘤进行有效杀伤。为了能克服这一问题,新一代car-t细胞技术应运而生,这种被改造后的t细胞在原来第二代和第三代car-t细胞的基础型上,引入可以辅助靶向识别、促进原位炎症反应或者封闭免疫检测点信号等功能分子,从而起到协同并提高car-t细胞的肿瘤靶向性和抗肿瘤杀伤活性(3)。
car-t细胞疗法的出现改变了手术、放疗、化疗、靶向疗法以及激素疗法等传统肿瘤治疗的理念,其可靶向抗原的广谱性和多样性,以及在体内代谢增值的可持续性等特点,为肿瘤的治疗提供了更多更有效的选择。但是作为一种“活的”药物,car-t与其他治疗方法有很大区别,其潜在安全性问题不容忽视,比较常见的包括靶向毒性、脱靶效应、细胞因子释放综合征等(4)。
在理想状态下,car-t细胞的靶点应该是对肿瘤细胞存活不可或缺的肿瘤特异性表面标志物,但实际上很多car-t细胞的靶点在正常组织细胞上也有表达(以相等或较低密度),当这样的分子被用作car-t细胞的靶点时,正常细胞也会受到car-t细胞的攻击而损伤,严重时可导致患者死亡,这就是所谓的靶向效应(on-target/off-tumor)(5,6)。典型的car-t治疗引起致命不良反应的例子为morgan等报道的,用针对人类表皮生长因子受体ii(her2)设计的第三代car-t细胞治疗一名结直肠癌转移的患者,在静脉注射car-t细胞4天后患者死亡;经尸检发现,死亡是由于肺上皮细胞表面低水平表达her2,而回输的car-t细胞攻击了患者的肺部,导致肺毒性引起的(7)。为了克服靶向效应的问题,选择合适的靶点抗原至关重要。除了确认靶点主要存在于癌变细胞上,还应考虑到由于照射、细胞凋亡或car-t细胞的扩增和激活等因素可能导致正常和癌变细胞上的靶蛋白表达水平发生改变。另外,细胞因子释放所造成的细胞因子释放综合症(crs)是在car-t注射及预处理过程中,机体免疫细胞大量活化、溶解,导致细胞因子被大量释放,从而引起全身免疫风暴,是目前最常见的car-t细胞注射后免疫反应副作用,同时也是car-t细胞在体内“发挥作用”的标志。crs会导致患者血液中细胞因子水平显著上升,可能出现发烧、头痛、恶心、腹泻、心动过速、低血压、缺氧、癫痫发作、精神混乱等症状,严重时可致多器官衰竭和死亡。crs中细胞因子大量释放的机制尚不明确,可能包括过度活化的car-t细胞、死亡的肿瘤细胞、活化的巨噬细胞(以及其他先天免疫细胞)和血管内皮细胞等(8,9)。因此在考虑car-t细胞免疫疗效的同时,如何保证患者的安全是目前car-t细胞免疫疗法研究的热点。
除了有效的临床干预之外,科学家更注重car-t本身的技术改造,包括可拆分car-t技术,自杀基因调控car-t细胞,synnotch受体技术,肿瘤微环境驱动型car-t细胞以及肿瘤抗原特异性car-t细胞等技术的开发为提高car-t临床安全性方面提供了很好的借鉴。此外,可调控car-t细胞作为一种独特的car-t技术被一些实验室相继报道,该技术采用半抗原小分子修饰肿瘤靶向抗体用来制备调控分子,进一步结合半抗原特异性的car-t细胞用于血液癌和实体瘤的免疫治疗(10)。可调控car-t技术在解决安全性的方面提出了新的理念,通过调控分子来启动car-t细胞的抗肿瘤活性和活化增殖,这种将调控分子与car-免疫细胞分开构建的思路及方法,能够有效地解决car-t细胞注射后所产生的安全性问题。传统的可调控car-t技术策略依赖于单靶点识别调控分子,或者采用两个不同的单靶点调控分子协同作用介导car-t细胞的抗肿瘤功能。
目前,car-t临床试验研究主要集中在血液系统肿瘤和实体肿瘤方面,特别是在血液系统肿瘤如急性白血病和非霍奇金淋巴瘤的治疗上已经取得巨大突破。然而实体肿瘤存在于肿瘤微环境中,肿瘤组织内细胞组成复杂,不同的肿瘤细胞有不同的蛋白表达谱,单一靶点难以覆盖所有的肿瘤细胞,因此针对实体肿瘤的car-t疗法进展缓慢。根据已有的理论与实践经验,即使表达某种抗原的肿瘤细胞能够被有效清除,患者体内仍然会残留大量的抗原阴性肿瘤细胞,这种不完全清除无法达到肿瘤治愈的要求,是影响car-t免疫治疗效果的重要因素之一。另外,一些研究表明接受传统的car-t细胞疗法的患者中出现了癌症再度复发的情况,其复发机制在于肿瘤细胞自身的抗原调节现象导致抗原靶向表位出现变化或抗原表达水平减弱造成免疫逃逸,导致car-t治疗的失效。由于治疗诱发肿瘤细胞目的抗原自身的修饰、表达量的减少或者性质改变从而避免免疫细胞的杀伤,也是目前影响car-t免疫治疗效果的另一重要因素之一。针对肿瘤免疫逃逸现象目前已有两种应对方案,一是实现针对两种不同肿瘤靶点的car在t细胞共表达;二是设计双靶点识别特异性的单一car靶向策略,这些串联的car包含两个不同的抗原结合结构域。但无论系统设计如何,在这两种情况下,至少一种表面靶点的存在足以激活car-t细胞并发起细胞裂解,从而降低反应性肿瘤生长的机会,或者尽可能延迟其生长。双靶点car-t细胞联合可寻求更高比例肿瘤细胞的靶向覆盖,提高car-t细胞杀伤肿瘤细胞的比例,从而可以提高患者的生存获益(11,12)。
传统双靶点car-t细胞在提高抗肿瘤功效,避免免疫逃逸的同时,往往会带来意想不到的问题。第一种策略因为需要在t细胞中同时引入带有双car的大片段基因,通常会造成car的不对等表达或者低表达,而通过串联抗体引入的双特异性car,在制备过程中通常因为串联表达的抗体在细胞表面的错配或者空间位阻造成car-免疫细胞靶向识别功能降低,甚至造成严重的脱靶毒性。
目前,针对实体肿瘤的理想靶标较少,作为人类表皮细胞生长因子受体家族成员,her2在正常组织中低表达或者不表达,而在多种肿瘤(包括乳腺癌、肺癌、前列腺癌、头颈部鳞癌、胃肠道肿瘤和妇科肿瘤)中都有高表达,因此是car-t治疗的潜在靶点。人类表皮生长因子受体2也称之为erbb-2、c-erbb2或her2/neu。her2基因定位于染色体17q21全长28515bp,转录4624nt的mrna,编码一个185kda蛋白(p185)。her2作为一个跨膜蛋白包含3个区域:n端胞外结构域,含一个α螺旋的跨膜区和细胞内的酪氨酸激酶结构域。胞外区(氨基酸22~653)可分为4个亚结构域(ⅰ~ⅳ),ⅰ、ⅲ亚结构域可以作为配体的结合位点,ⅱ、ⅳ亚结构域由于存在丰富的半胱氨酸,可以形成同源或异源二聚体。跨膜区是由23个氨基酸组成的单一α螺旋结构。胞内区(氨基酸676~1259)包含了多个重要的环状结构,构成了酪氨酸激酶的活性位点(13)。大约15%至20%的乳腺癌肿瘤细胞表面显示出her2蛋白的过度表达,卵巢癌中her2的过表达与乳腺癌中相似,占15%~30%。her2的阳性表达多与淋巴结转移、较大的肿瘤直径、较高的恶性程度、低表达的雌、孕激素受体状态、绝经后状态以及较短的总生存期和无病生存期显著相关。研究表明her2的过表达可以异常激活多条信号传导通路从而导致肿瘤细胞的过度增殖和侵袭,其中包括pi3k和mapk等,另有研究表明her2的活化能够导致脂肪酸合酶过表达且诱导多种肿瘤细胞的恶性表型,脂肪酸合酶介导的脂肪酸从头合成在多种肿瘤的形成过程中具有重要作用。因此抑制肿瘤细胞表面her2的过表达及其生物学功能具有积极意义。2002年9月第一个靶向her2的人源化单克隆抗体曲妥珠单抗在中国获批上市,自曲妥珠单抗问世以来不仅明显改善了乳腺癌患者预后情况,也极大地提高了乳腺癌辅助化疗患者的无病生存率和总生存率。作为首个靶向抗her2的人源化单克隆抗体,曲妥珠单抗影响了乳腺癌的诊治模式,是乳腺癌药物治疗的重大突破。2013年2月一种靶向her2的单克隆抗体和微管抑制剂偶联物ado-trastuzumabemtansine在美国获批。2018年12月国家食药局批准帕托珠单抗上市,帕托珠单抗是一种靶向her2的人源化单克隆抗体,通过抑制her2及其家族成员之间的配体依赖性异二聚化作用,阻断细胞周期并诱导其凋亡。另外,拉帕替尼、吡咯替尼、来那替尼等靶向her2或共靶向her2、egfr等受体的药物已在临床应用中取得良好结果。然而,上述获批的药物目前只是用于her2高表达的肿瘤患者,而对于her2低表达或者抗原异质性,以及靶向治疗造成的her2抗原逃逸等病人,其预后较差。有研究表明大约有30%her2过表达的肿瘤存在p95her2剪切体,该剪切体是her2阳性肿瘤的预后标志物,从而对已经上市的her2靶向药物包括曲妥珠单抗、帕托珠单抗和ado-trastuzumabemtansine的治疗产生耐药性,这种耐药性与her2表达阴性肿瘤细胞所表现的耐药性相似。此外,虽然目前已经有her2靶向的car-t细胞产品用于临床前和临床肿瘤病人的治疗,但在接受传统的car-t治疗之后,her2阳性细胞抗原发生突变,使car-t细胞无法成功识别肿瘤细胞,造成免疫逃逸,类似抗原修饰的问题还包括her2在肿瘤表面表达水平降低现象(14)。另一方面,her2作为car-t细胞疗法的靶点,由于其组织靶向毒性,会引起严重的呼吸窘迫及肺部浸润等毒副作用,从而造成患者死亡(7)。这些情况充分印证了her2靶向car-t疗法带来的治疗有效性及靶向安全性问题,因此寻求her2以外的另外靶点用于辅助治疗,并结合可调控开关策略,可以有效地解决her2单靶点传统car-t细胞治疗带来的潜在问题。
有研究报道,肿瘤细胞表面的her2受体可以和包括其它her家族受体、met以及胰岛素样生长因子1受体(insulin-likegrowthfactor1receptor,igf1r)等受体相互作用,影响下游的信号转导功能。其中igf1r受体在急性白血病、乳腺癌等多种恶性肿瘤中均有异常表达,多种证据表明igf1r信号转导通路通过多种机制介导而参与了肿瘤的发生、有丝分裂、转移、血管发生和抗凋亡等过程、并涉及肿瘤的耐药、放疗抵抗及her2靶向治疗。igf1r与her2的协同作用,可以引发严重的肿瘤侵袭以及潜在的her2靶向治疗的耐药性。正是由于her2和igf1r之间的这种相互关系,靶向这两个酪氨酸激酶受体的联合治疗策略应运而生。目前已有报道,her2/igf1r联合靶向治疗的抗体和小分子抑制剂,在临床前的研究中显示了良好的抗肿瘤效果。her2/igf1r双靶向药物在乳腺癌中显示出显著的抗肿瘤活性,受体特异性更广的药物可能比单受体选择性药物更有效。关于乳腺癌mcf-7细胞的研究表明,尽管her2靶向抗体曲妥珠单抗和igf1r小分子拮抗剂各自仅对细胞周期的影响微乎其微,但它们的组合可以最大程度上的抑制受体下游的erk和akt磷酸化,可以显著增强其肿瘤抑制效果(15)。与此同时,有研究显示her2与igf1r靶向药物neratinib和bms-754807的单独使用均不能够诱导三阴性乳腺癌细胞的凋亡,但是两种受体拮抗剂的联合可以诱导显著的细胞凋亡(16)。此外,有研究将携带有igf1r受体或者异构体的dna疫苗注射入her2阳性并可产生il-12的肿瘤细胞,用于制备辅助型细胞疫苗,其临床前的实验结果显示,该双靶点疫苗可以显著地诱导igf1r抗体的表达,从而显著地延缓自发性横纹肌肉瘤的发生(17)。虽然her2/igf1r双靶点抑制策略已经从分子机理等方面得到了详细的阐述,然而目前还没有将her2/igf1r协同靶向的思路拓展到免疫治疗甚至car-t治疗策略中,其主要的考虑在于igf1r表达的广谱性,以her2/igf1r为靶点的car-t细胞会潜在地引起机体靶向毒性。
参考文献
1.mellmani,coukosg,dranoffg.cancerimmunotherapycomesofage.nature.2011;480(7378):480-9.
2.grossg,wakst,eshharz.expressionofimmunoglobulin-t-cellreceptorchimericmoleculesasfunctionalreceptorswithantibody-typespecificity.procnatlacadsciusa.1989;86(24):10024-8.
3.rodgersdt,mazagovam,hamptonen,caoy,ramadossns,hardyir,etal.switch-mediatedactivationandretargetingofcar-tcellsforb-cellmalignancies.procnatlacadsciusa.2016;113(4):e459-68.
4.pageljm,westhj.chimericantigenreceptor(car)t-celltherapy.jamaoncol.2017;3(11):1595.
5.casuccim,hawkinsre,dottig,bondanzaa.overcomingthetoxicityhurdlesofgeneticallytargetedtcells.cancerimmunolimmunother.2015;64(1):123-30.
6.dottig,gottschalks,savoldob,brennermk.designanddevelopmentoftherapiesusingchimericantigenreceptor-expressingtcells.immunolrev.2014;257(1):107-26.
7.morganra,yangjc,kitanom,dudleyme,laurencotcm,rosenbergsa.casereportofaseriousadverseeventfollowingtheadministrationoftcellstransducedwithachimericantigenreceptorrecognizingerbb2.molther.2010;18(4):843-51.
8.minagawak,zhoux,mineishis,distasia.seatbeltsincartherapy:howsafearecars?pharmaceuticals(basel).2015;8(2):230-49.
9.mausmv,gruppsa,porterdl,junech.antibody-modifiedtcells:carstakethefrontseatforhematologicmalignancies.blood.2014;123(17):2625-35.
10.caoy,rodgersdt,duj,ahmadi,hamptonen,majs,etal.designofswitchablechimericantigenreceptortcellstargetingbreastcancer.angewchemintedengl.2016;55(26):7520-4.
11.majs,kimjy,kazanesa,choish,yunhy,kimms,etal.versatilestrategyforcontrollingthespecificityandactivityofengineeredtcells.procnatlacadsciusa.2016;113(4):e450-8.
12.kimms,majs,yunh,caoy,kimjy,chiv,etal.redirectionofgeneticallyengineeredcar-tcellsusingbifunctionalsmallmolecules.jamchemsoc.2015;137(8):2832-5.
13.chohs,masonk,ramyarkx,stanleyam,gabellisb,denneydw,jr.,etal.structureoftheextracellularregionofher2aloneandincomplexwiththeherceptinfab.nature.2003;421(6924):756-60.
14.majznerrg,mackallcl.tumorantigenescapefromcart-celltherapy.cancerdiscov.2018;8(10):1219-26.
15.sanabria-figueroae,donnellysm,foykc,bussmc,castellinorc,paplomatae,etal.insulin-likegrowthfactor-1receptorsignalingincreasestheinvasivepotentialofhumanepidermalgrowthfactorreceptor2-overexpressingbreastcancercellsviasrc-focaladhesionkinaseandforkheadboxproteinm1.molpharmacol.2015;87(2):150-61.
16.chakrabortya,hatzisc,digiovannamp.co-targetingtheherandigf/insulinreceptoraxisinbreastcancer,withtripletargetingwithendocrinetherapyforhormone-sensitivedisease.breastcancerrestreat.2017;163(1):37-50.17.degiovannic,landuzzil,palladinia,ianzanoml,nicolettig,ruzzif,etal.cancervaccinesco-targetingher2/neuandigf1r.cancers(basel).2019;11(4).
技术实现要素:
本发明的目的在于解决car-t治疗在临床应用中出现的有效性和安全性问题中的至少一个。
本发明所采取的技术方案是:
发明人将能够靶向肿瘤细胞表面不同受体的双靶点识别分子与car-免疫细胞的活化结构域分开构建,在肿瘤靶向治疗中,采用标签分子修饰的双靶点识别分子作为调控分子,通过在免疫细胞上工程化修饰标签特异性car,制备新型的双靶点识别可调控基因工程化免疫细胞。其中调控分子不仅能够与带有特异性标签分子识别蛋白的car胞外识别结构域结合,实现对car-t细胞的活化,从而保证car-t细胞在患者体内的可控性,还可以同时靶向肿瘤细胞表面的不同抗原分子以提高car-t细胞对肿瘤的识别效率,从而克服传统car-t细胞的单靶点识别造成的免疫逃逸和肿瘤复发等问题。该car-免疫细胞及双靶点调控分子的组合物,在实现对肿瘤不同抗原多靶点识别,避免单靶点治疗造成的免疫逃逸的同时,提高其安全性,并有效解决car-t细胞治疗过程中潜在的脱靶毒性等问题。
一种可调控car-免疫细胞的组合物,包括car-免疫细胞和肿瘤特异性靶点识别分子,所述car-免疫细胞的car胞外识别结构域连接有标签分子特异性识别分子;
所述肿瘤特异性靶点识别分子具有至少2个可识别肿瘤细胞表面不同抗原的识别分子区和标签分子区,所述标签分子区可与所述car-免疫细胞的car胞外识别结构域的所述标签分子特异性识别分子特异性结合。
标签分子天然不存在于人体内,因此,在没有外源标签分子的情况下,car胞外识别结构域连接有标签分子特异性识别分子的car-免疫细胞不会和体内的细胞发生作用,避免了car-免疫细胞带来的潜在毒性。
在本发明的一些组合物实例中,所述car-免疫细胞中的标签特异性识别分子选自单链抗体、蛋白配体、短肽分子或者核酸序列。
在本发明的一些组合物实例中,所述标签分子选自fitc,生物素,his标签,flag标签,myc标签,gcn4标签,ha标签,v5标签,壳聚糖结合蛋白(cbp)标签,麦芽糖结合蛋白(mbp)标签和谷胱甘肽-s-转移酶(gst)标签中的一种。优选易于生产、纯化且免疫原性低的标签分子。
在本发明的一些组合物实例中,所述肿瘤特异性靶点识别分子的识别分子区和标签分子区直接偶联或通过支架蛋白偶联在一起,所述支架蛋白包括adnectin、亲和体(affibody)、anticalin、bicyclicpeptide、darpin、fynomer、kunitzdomain、e7免疫蛋白(im7)、淋巴细胞受体可变区(vlr)、单域抗体、全抗体、抗体片段,以及dnaaptamer。
在本发明的一些组合物实例中,所述肿瘤特异性靶点识别分子识别的抗原选自her2、igf1r、egfr、her3、her4、vegfr1/2、c-met、muc-1、il13r;优选的,所述肿瘤特异性靶点识别分子识别的两种抗原为细胞表面her2受体的胞外结合域和igf1r受体的胞外结合域。
在本发明的一些组合物实例中,识别her2受体的胞外结合域的抗体分子是4d5;进一步的,识别igf1r受体的胞外结合域的抗体分子是人纤连蛋白衍生物adnectin。
在本发明的一些组合物实例中,所述的标签分子为myc标签,优选的,所述肿瘤特异性靶点识别分子为4d5/myc/adnectin,进一步的,所述4d5/myc/adnectin的蛋白序列为:
diqmtqspsslsasvgdrvtitcrasqdvntavawyqqkpgkapklliysasflysgvpsrfsgsrsgtdftltisslqpedfatyycqqhyttpptfgqgtkveikrtggggsggggsggggsevqlvesggglvqpggslrlscaasgfnikdtyihwvrqapgkglewvariyptngytryadsvkgrftisadtskntaylqmnslraedtavyycsrwggdgfyamdywgqgtlvtvssaaaggggsggggsggggseqkliseedlgstsgsgkpgsgegstkgdivsdvprdlevvaatptslliswsarlkvaryyritygetggnspvqeftvpknvytatisglkpgvdytitvyavtkmrdyrpisinyrteidkpsq。
在本发明的一些组合物实例中,所述肿瘤特异性靶点识别分子的识别分子区和标签分子区之间通过亲水性双功能连接子偶联,优选的,所述亲水性双功能连接子为连接肽,进一步的,所述连接肽选自线性多肽、β-折叠肽或卷曲螺旋肽。
在本发明的一些组合物实例中,所述肿瘤特异性靶点识别分子通过基因重组的方式融合表达,或通过随机化学偶联以及位点特异性化学键合的方式制备。
在本发明的一些组合物实例中,所述免疫细胞选自t细胞,自然杀伤细胞,巨噬细胞,肿瘤免疫细胞以及干细胞诱导的免疫细胞。t细胞的类型没有特殊要求,优选的,t细胞选自cd4、cd8、treg、th17、th9、tfr、tfh、tfc、tph、trm、初始t细胞、中央记忆t细胞、效应记忆t细胞、效应t细胞、pro、pre、成熟t细胞。
在本发明的一些组合物实例中,car-免疫细胞按现有的方法构建得到,包括但不限于慢病毒、腺病毒、腺相关病毒、转座子技术、mrna转染、纳米基因,zinefingernuclease,talen以及crispr/cas9。
在本发明的一些组合物实例中,car结构中的铰链区选自免疫球蛋白家族或者肿瘤坏死因子超家族中的至少一种。
在本发明的一些组合物实例中,car-免疫细胞的car结构中还包括协同刺激因子cd28,4-1bb、ox-40、cd27,icos中的至少一种。
本发明的有益效果是:
本发明的一些实例,通过将靶向肿瘤表面不同受体的识别分子与标签分子进行连接,构建双靶点识别调控分子,从而介导靶向标签分子的car-免疫细胞对肿瘤细胞的双靶点识别杀伤作用。通过控制双靶点识别调控分子的添加量,可以有效调控car-免疫细胞对肿瘤细胞的识别杀伤作用,具有更好的安全性。
本发明的一些实例,双靶点识别调控分子拥有同时识别肿瘤细胞表面的两个不同抗原的识别结构域,其中单链抗体4d5识别肿瘤细胞表面her2抗原,adnectin识别肿瘤细胞表面igf1r抗原,从而使一个调控分子具备同时识别肿瘤细胞两个靶点的功能,识别分子特异性中肿瘤特异性识别部分还能够替换为adnectin、亲和体(affibody)、anticalin、bicyclicpeptide、darpin、fynomer、kunitzdomain、e7免疫蛋白(im7)、淋巴细胞受体可变区(vlr)、单域抗体、全抗体和抗体片段,以及dnaaptamer等多种支架蛋白所构成的分子来靶向不同的肿瘤表面抗原;此外,fitc,生物素,his标签,flag标签,myc标签,gcn4标签,ha标签,v5标签,壳聚糖结合蛋白(cbp)标签,麦芽糖结合蛋白(mbp)标签和谷胱甘肽-s-转移酶(gst)标签等都可作为介导car胞外识别结构域相连的标签分子以构建双靶点调控分子。本发明所构建的双靶点肿瘤特异性识别分子能够降低肿瘤抗原免疫逃逸以及治疗中靶向和脱靶毒性的潜在风险,在一定程度上提高了car-t细胞治疗的有效性和安全性。
附图说明
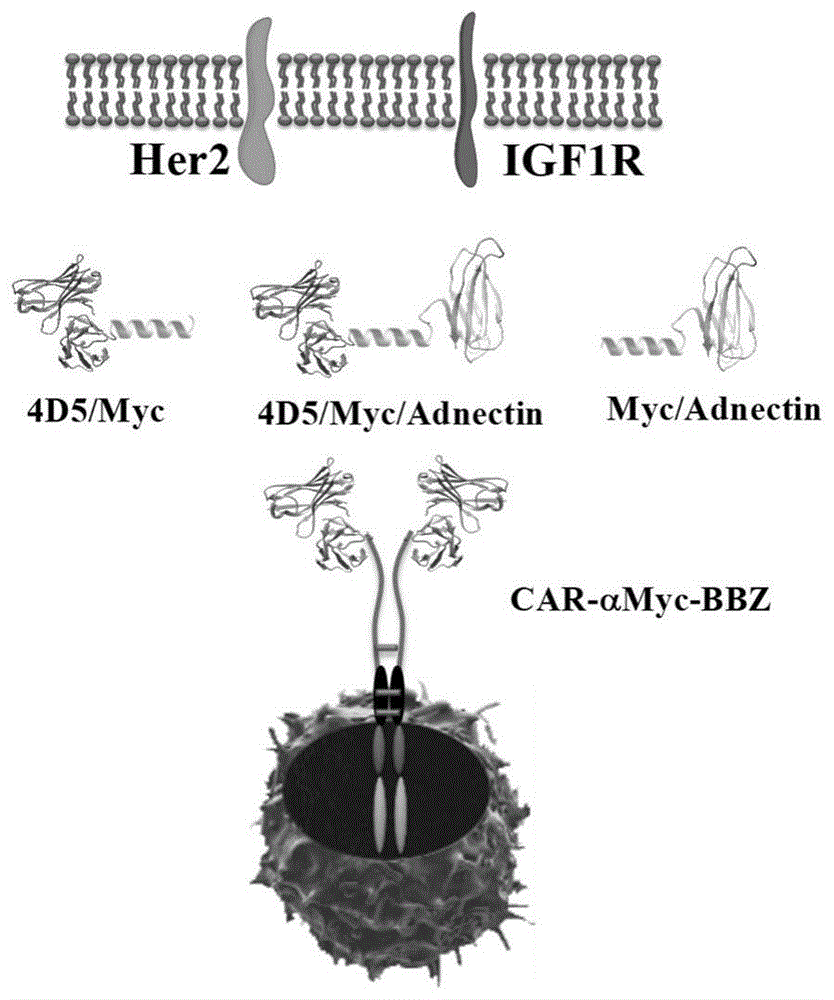
图1是不同调控分子与car-t细胞组合与肿瘤表面受体的示意图;
图2是肿瘤识别调控分子的结构示意图;
图3是采用凝血酶对不同肿瘤识别调控分子的酶切电泳及免疫印迹验证结果;
图4是myc标签靶向car的结构示意图;
图5是不同调控分子与不同her2和igf1r表达水平的肿瘤细胞结合活性的流式细胞仪分析结果;
图6是不同调控分子介导car-t细胞对不同her2和igf1r表达水平肿瘤细胞的杀伤结果;
图7~10是不同调控分子介导car-t细胞对肿瘤杀伤的car-t活化能力分析;
图11~13是不同调控分子介导car-t细胞对肿瘤杀伤的细胞因子释放分析。
具体实施方式
下面结合示例性的实验,进一步说明本发明的技术方案。
本发明以恶性肿瘤相关抗原her2和igf1r的胞外结构域作为识别靶点,构建可识别两种抗原蛋白的双靶点调控分子,进一步引入标签分子和标签靶向的car-免疫细胞,制备双靶点识别的可调控car-免疫细胞策略(图1)。
一、调控分子的构建
1.调控分子表达载体的构建
采用生物融合的方法制备调控分子,表达结构如图2所示。将4d5/myc、myc/adnectin以及4d5/myc/adnectin的目的基因通过pcr的方法在n端引入his标签和凝血酶切位点,经过msci和xhoi双酶切后,采用t4dna连接酶克隆入pet32a(+)载体上,转化至dh5α感受态细胞后涂板,并挑取单克隆进行测序鉴定。
其中,4d5/myc的蛋白序列为:
diqmtqspsslsasvgdrvtitcrasqdvntavawyqqkpgkapklliysasflysgvpsrfsgsrsgtdftltisslqpedfatyycqqhyttpptfgqgtkveikrtggggsggggsggggsevqlvesggglvqpggslrlscaasgfnikdtyihwvrqapgkglewvariyptngytryadsvkgrftisadtskntaylqmnslraedtavyycsrwggdgfyamdywgqgtlvtvssaaaggggsggggsggggseqkliseedl(seqidno.:1)
myc/adnectin的蛋白序列为:
eqkliseedlgstsgsgkpgsgegstkgdivsdvprdlevvaatptslliswsarlkvaryyritygetggnspvqeftvpknvytatisglkpgvdytitvyavtkmrdyrpisinyrteidkpsq(seqidno.:2)
4d5/myc/adnectin的蛋白序列为:
diqmtqspsslsasvgdrvtitcrasqdvntavawyqqkpgkapklliysasflysgvpsrfsgsrsgtdftltisslqpedfatyycqqhyttpptfgqgtkveikrtggggsggggsggggsevqlvesggglvqpggslrlscaasgfnikdtyihwvrqapgkglewvariyptngytryadsvkgrftisadtskntaylqmnslraedtavyycsrwggdgfyamdywgqgtlvtvssaaaggggsggggsggggseqkliseedlgstsgsgkpgsgegstkgdivsdvprdlevvaatptslliswsarlkvaryyritygetggnspvqeftvpknvytatisglkpgvdytitvyavtkmrdyrpisinyrteidkpsq(seqidno.:3)
2.调控分子的表达与纯化
将构建的质粒转染至bl21、rosettagami(de3)plyss或者tuner(de3)plyss,涂板后挑取单克隆过夜培养,按1:100接种于500ml新鲜tb培养基内直至od600值达到1.0~1.2时,iptg诱导20小时;4℃下离心收集菌体,称重后于-80℃冻存。加入菌体重量10倍体积的裂解液,然后超声破碎细胞,至菌液不再粘稠。将细胞裂解液4℃离心1小时后,将上清液采用0.45μm滤膜过滤。将菌体裂解上清液采用镍柱纯化,选择洗涤缓冲液冲洗镍柱上的杂蛋白。再用洗脱缓冲液洗脱目的蛋白。将洗脱下来的目的蛋白采用tris-hcl/nacl缓冲液透析后,加入凝血酶室温酶切16小时,通过sds-page电泳验证酶切完全后(图3),采用镍树脂去除酶切标签。最后使用内毒素去除树脂填料去除内毒素,之后用内毒素检测试剂盒(genscript,l00350)检测内毒素含量,保证内毒素含量在5eu/kg以下。
3.sds-page及westernblot验证融合蛋白
取适量纯化后的蛋白分子,加入蛋白样品体积三分之一的还原上样缓冲液,涡旋混匀后离心。金属浴煮样10分钟后离心。采用12%分离胶,5%的浓缩胶,将凝胶板固定于模具中,倒入电泳缓冲液观察是否漏液,拔去梳子,依次上样;上样缓冲液补充至刻度线,电压调至80v开始电泳,待样品进入分离胶后将电压调成120v,观察电泳情况;将膜、滤纸、海绵在使用前浸泡在电转缓冲液中,一块胶按照白板-海绵-滤纸-膜-胶-滤纸-海绵-黑板的顺序依次放好(确保无气泡),放置在冰上,连接电源220ma,2h。另一块胶使用考马斯亮蓝染色,清水脱色观察蛋白电泳情况。转膜结束后将膜取出,裁剪成适当大小,标记蛋白分子量标准品。加入封闭液(5%脱脂奶粉+0.12%tween20tbst),室温封闭1h。tbst震荡清洗3分钟。室温震荡孵育一抗2h。用适量tbst漂洗nc膜10分钟/次,共3次。室温震荡孵育二抗1h,用适量tbst漂洗10分钟/次,共3次。凝胶成像仪显影。结果如图3所示,4d5/myc、myc/adnectin和4d5/myc/adnectin在凝血酶酶切前后,显示了正确的分子量以及较好的纯度,免疫印迹进一步对其标签myc蛋白进行了验证。
二、car-t细胞的制备
1.载体构建
以慢病毒载体pelps构建靶向myc的car为例(图4),将myc靶向抗体9e10为例,将抗体的可变区采用vl-连接肽-vh和vh-连接肽-vl的方式,引入二代car的结构中,常规的二代car结构包括ef1α启动子,信号肽分子,单链抗体,铰链区,跨膜区,共刺激分子以及cd3zeta结构域。将构建的car采用bamhi和sali双酶切后使用t4dna连接酶克隆至pelps载体,或者采用同源重组试剂盒构建载体。
代表性的car结构如下:
pelps-car-anti-myc9e10(vl-vh)-bbz蛋白序列:
malpvtalllplalllhaarpdivltqspaslavslgqratiscrasesvdnygfsfmnwfqqkpgqppklliyaisnrgsgvparfsgsgsgtdfslnihpveeddpamyfcqqtkevpwtfgggtkleikggggsggggsggggsevhlvesggdlvkpggslklscaasgftfshygmswvrqtpdkrlewvatigsrgtythypdsvkgrftisrdndknalylqmnslksedtamyycarrsefyyygntyyysamdywgqgasvtvsstttpaprpptpaptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgrkkllyifkqpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapaykqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr(seqidno.:4)
pelps-car-anti-myc9e10(vh-vl)-bbz蛋白序列:
malpvtalllplalllhaarpevhlvesggdlvkpggslklscaasgftfshygmswvrqtpdkrlewvatigsrgtythypdsvkgrftisrdndknalylqmnslksedtamyycarrsefyyygntyyysamdywgqgasvtvssggggsggggsggggsdivltqspaslavslgqratiscrasesvdnygfsfmnwfqqkpgqppklliyaisnrgsgvparfsgsgsgtdfslnihpveeddpamyfcqqtkevpwtfgggtkleiktttpaprpptpaptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgrkkllyifkqpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapaykqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr(seqidno.:5)
2.慢病毒的制备
将293ft细胞培养于dmem培养基(10%fbs,1%青霉素链霉素双抗,1%非必需氨基酸,1%丙酮酸钠,1%l-谷氨酰胺),待细胞贴壁后密度达到80%左右时,提前1h更换无抗生素dmem培养基。质粒按照pelps-car:pmdlgprre:prsv-revp:md2.g=2.25:1.8:1.8:0.75(μg)比例混合于0.3mlopti-mem中,将20μllipofectamine2000加入0.3mlopti-mem中,静置5分钟后将二者混合并涡旋使其混合均匀,室温下静置30分钟后垂直顺时针滴加到293ft细胞中。细胞在恒温培养箱中培养6小时后,更换新的无抗生素dmem培养基,37℃,5%co2再培养48小时。收集上清液,4℃,1500rpm离心10分钟,弃细胞沉淀,上清即为包装好的病毒,-80℃保存备用。
3.人pbmc(peripheralbloodmononuclearcell,外周血单核细胞)的提取
取15mlficoll-poque于离心管内(血液与ficoll-poque比例以2:1为宜)。将取自健康志愿者的新鲜血液缓慢倾斜加入到装有ficoll-poque的离心管内,使细胞悬液重悬于分层液上。用水平离心机室温,2000rpm离心30分钟(升速为9,降速为0)。离心结束后取出离心管会观察到分为4层:最上层是血浆,含部分血小板;第二层为薄薄的白膜层,主要是单个核细胞,还混杂有少量血小板;第三层为分离液层;第四层为粒细胞及红细胞,红细胞沉降于管底,而粒细胞则紧贴在红细胞上呈一层很薄的白膜,即pbmc。用巴氏吸管吸去上层血清(可离心后分装保存),再小心吸取白膜状分层,加入到两只离心管内(体积尽量相同)。加入rpmi-1640培养基至50ml处,上下颠倒混匀。水平离心机27℃,2000rpm离心10分钟(升速为9,降速为9),低速离心有利于去除细胞悬液中残留的血小板,离心后去上清。加入aim-v培养基27℃,2000rpm再次离心10分钟,离心后去上清。用15mlaim-v培养基重悬后加入到t75培养瓶内。37℃培养1h以上,此步骤目的在于去除贴壁细胞。之后吸取上清至离心管内,加入新鲜rpmi-1640培养基37℃恒温培养,待用。
4.pbmc冻存
1×108个pbmc细胞重悬至700μl冻存液中,重悬液转移至冻存管内,冻存管放入程序降温盒内,将程序降温盒转移至-80℃超低温冰箱,24h以后转移至液氮长期保存。
5.t细胞的激活
将从液氮中取出冻存的pbmc迅速置于37℃水浴快速融化,加入10mlaim-v培养基24℃,1500rpm离心5分钟。离心后吸去上清,再用适量aim-v培养基重悬(若有颗粒,可转移上清)。按照每106个细胞30μl磁珠(用前涡旋)的比例取适量磁珠,用2~3ml含5%fbs的pbs洗磁珠(磁珠会吸附在侧壁)。吸去磁珠的上清后,再用aim-v培养基洗一遍。t细胞计数后用培养基稀释成1×106/ml后加入磁珠悬液。加入il-2,il-2起始浓度为2.6×105iu/ml,调至终浓度为300iu/ml。
6.t细胞的慢病毒转导
将活化24h的t细胞,采用1500rpm离心5分钟,进一步使用rpmi-1640培养基重悬,使用血球计数板和台盼蓝对其进行细胞计数,将合适数量的细胞悬液与病毒溶液混合后加入10mg/ml的polybrene(聚凝胺),使其终浓度为8μg/ml以及300iu/ml的il-2。于33℃,1000g离心90分钟,于37℃恒温培养箱中过夜培养。第二天将细胞离心后补充新鲜培养液37℃培养箱继续培养,待表达稳定后用于抗肿瘤活性检测。
三、功能验证
1.调控分子的结合活性
收集不同her2和igf1r表达水平的的乳腺癌肿瘤细胞,采用200nm调控分子(4d5/myc、myc/adnectin和4d5/myc/adnectin),4℃孵育2小时后,使用facs缓冲液清洗3次后,采用apc-抗his标签二抗4℃孵育1小时,采用流式细胞分析仪进行分析。结果如图5所示,结果显示,4d5/myc、myc/adnectin、4d5/myc/adnectin保留了对不同her2和igf1r表达肿瘤细胞的靶向识别能力,尤其是对her2高表达乳腺癌细胞的识别能力较强,该识别能力主要依赖于4d5对her2靶点结合功能,并辅以igf1r的结合。
2.肿瘤细胞裂解实验
分别取1×105个skbr3、mda-mb-453、mda-mb-435、mda-mb-231、mcf-7、mda-mb-468细胞加入96孔板中,设置阴性对照组(肿瘤细胞+car-t细胞)和阳性对照组(肿瘤细胞+细胞裂解液)。调节car-t细胞与肿瘤细胞的比例为10:1,将car-t细胞分别加入96孔板对应孔中。分别加入系列稀释的4d5/myc、myc/adnectin、4d5/myc+myc/adnectin、4d5/myc/adnectin之后,37℃,5%co2培养箱培养24h,离心收集上清液,采用promega的非放射性细胞毒性检测试剂盒进行检测,采用graphpadprism作图分析实验结果如图6,虽然4d5/myc和myc/adnectin均能有效地杀伤肿瘤细胞,但双靶点调控分子4d5/myc/adnectin不仅对高表达her2的肿瘤细胞展现了有力的杀伤效果,在介导car-t细胞对her2低表达肿瘤细胞的杀伤作用中依然能保持良好的杀伤效果,显著增强介导car-t细胞对肿瘤杀伤的广谱性和有效性,而且双靶点调控分子的活性显著高于两个单靶点调控分子作用效果的简单叠加,这得益于her2/igf1r共靶向所带来的高效性。
3.t细胞活性检测实验
分别取1×105个skbr3、mda-mb-453、mda-mb-231、mda-mb-468细胞加入96孔板中。调节car-t细胞与肿瘤细胞的比例为10:1,将car-t细胞分别加入96孔板对应孔中。分别加入10nm的4d5/myc、myc/adnectin、4d5/myc+myc/adnectin、4d5/myc/adnectin之后,37℃,5%co2培养箱培养一天。水平离心机离心,3000rpm,5分钟,去上清液,使用facs缓冲液清洗细胞。用facs缓冲液100倍稀释percp/cy5.5anti-humancd25抗体及apcanti-humancd69抗体配制染色液。96孔板每孔加入100μl染色液,冰上避光放置,染色1h。使用facs缓冲液清洗细胞两次并重悬至流式管中,流式细胞仪检测并分析car-t细胞活性。实验结果如图7~10所示,虽然4d5/myc和myc/adnectin均可介导car-t细胞对不同her2表达水平的肿瘤细胞的杀伤,并实现良好的抗原表达依赖的car-t细胞的活化,但是双靶点调控分子4d5/myc/adnectin在介导car-t细胞杀伤肿瘤的同时,显示了显著增强的car-t细胞活化增殖活性,而且双靶点调控分子介导car-t细胞的活化能力显著高于两个单靶点调控分子作用效果的简单叠加。
4.细胞因子释放测定
将1×105个肿瘤细胞接种到24孔板内,每个靶细胞设置两个复孔。car-t细胞使用pbs洗涤后,按照1×106/ml的细胞密度稀释到不含il-2的t细胞扩增培养基中,于1×106个t细胞接种于含有靶细胞的培养孔中。分别将10nm的4d5/myc、myc/adnectin、4d5/myc+myc/adnectin、4d5/myc/adnectin加入对应孔中,37℃,5%co2培养箱培养24h后,收集上清液,采用il-2、ifn-γ和tnf-αelisa试剂盒检测不同样品中il-2、ifn-γ和tnf-α的含量,结果如图11~13所示。上文所述,细胞因子分泌是car-t细胞发挥作用的标志。对细胞因子释放测定结果进行分析后,我们发现双靶点调控分子4d5/myc/adnectin在介导car-t细胞杀伤肿瘤的同时,与4d5/myc、myc/adnectin和4d5/myc+myc/adnectin相比显示了显著增强的细胞因子分泌,并且其对her2表达水平不同的乳腺癌细胞展现出了同样的显著增强效果。
综上,功能验证结果显示4d5/myc/adnectin双靶点识别分子介导的car-t细胞杀伤肿瘤效果从不同角度来看,都在安全性及有效性方面较传统的car-t疗法有较大提升。可调控分子与car-t细胞的组合极大程度上解决了传统car-t细胞在患者体内的不可控性,通过注射可调控分子来控制car-t细胞对肿瘤细胞的杀伤,提高了car-t疗法在使用过程中的安全性。同时,采用her2及igf1r双靶点识别策略有效避免了单靶点识别所存在的免疫逃逸问题,增加了car-t细胞对肿瘤细胞的有效杀伤。
sequencelisting
<110>北京大学深圳研究生院
<120>双靶点识别可调控工程化免疫细胞的制备方法
<130>
<160>3
<170>patentinversion3.5
<210>1
<211>272
<212>prt
<213>人工序列
<400>1
aspileglnmetthrglnserproserserleuseralaservalgly
151015
aspargvalthrilethrcysargalaserglnaspvalasnthrala
202530
valalatrptyrglnglnlysproglylysalaprolysleuleuile
354045
tyrseralaserpheleutyrserglyvalproserargphesergly
505560
serargserglythraspphethrleuthrileserserleuglnpro
65707580
gluaspphealathrtyrtyrcysglnglnhistyrthrthrpropro
859095
thrpheglyglnglythrlysvalgluilelysargthrglyglygly
100105110
glyserglyglyglyglyserglyglyglyglysergluvalglnleu
115120125
valgluserglyglyglyleuvalglnproglyglyserleuargleu
130135140
sercysalaalaserglypheasnilelysaspthrtyrilehistrp
145150155160
valargglnalaproglylysglyleuglutrpvalalaargiletyr
165170175
prothrasnglytyrthrargtyralaaspservallysglyargphe
180185190
thrileseralaaspthrserlysasnthralatyrleuglnmetasn
195200205
serleuargalagluaspthralavaltyrtyrcysserargtrpgly
210215220
glyaspglyphetyralametasptyrtrpglyglnglythrleuval
225230235240
thrvalserseralaalaalaglyglyglyglyserglyglyglygly
245250255
serglyglyglyglysergluglnlysleuileserglugluaspleu
260265270
<210>2
<211>127
<212>prt
<213>人工序列
<400>2
gluglnlysleuileserglugluaspleuglyserthrserglyser
151015
glylysproglyserglygluglyserthrlysglyaspilevalser
202530
aspvalproargaspleugluvalvalalaalathrprothrserleu
354045
leuilesertrpseralaargleulysvalalaargtyrtyrargile
505560
thrtyrglygluthrglyglyasnserprovalglngluphethrval
65707580
prolysasnvaltyrthralathrileserglyleulysproglyval
859095
asptyrthrilethrvaltyralavalthrlysmetargasptyrarg
100105110
proileserileasntyrargthrgluileasplysprosergln
115120125
<210>3
<211>389
<212>prt
<213>人工序列
<400>3
aspileglnmetthrglnserproserserleuseralaservalgly
151015
aspargvalthrilethrcysargalaserglnaspvalasnthrala
202530
valalatrptyrglnglnlysproglylysalaprolysleuleuile
354045
tyrseralaserpheleutyrserglyvalproserargphesergly
505560
serargserglythraspphethrleuthrileserserleuglnpro
65707580
gluaspphealathrtyrtyrcysglnglnhistyrthrthrpropro
859095
thrpheglyglnglythrlysvalgluilelysargthrglyglygly
100105110
glyserglyglyglyglyserglyglyglyglysergluvalglnleu
115120125
valgluserglyglyglyleuvalglnproglyglyserleuargleu
130135140
sercysalaalaserglypheasnilelysaspthrtyrilehistrp
145150155160
valargglnalaproglylysglyleuglutrpvalalaargiletyr
165170175
prothrasnglytyrthrargtyralaaspservallysglyargphe
180185190
thrileseralaaspthrserlysasnthralatyrleuglnmetasn
195200205
serleuargalagluaspthralavaltyrtyrcysserargtrpgly
210215220
glyaspglyphetyralametasptyrtrpglyglnglythrleuval
225230235240
thrvalserseralaalaalaglyglyglyglyserglyglyglygly
245250255
serglyglyglyglysergluglnlysleuileserglugluaspleu
260265270
glyserthrserglyserglylysproglyserglygluglyserthr
275280285
lysglyaspilevalseraspvalproargaspleugluvalvalala
290295300
alathrprothrserleuleuilesertrpseralaargleulysval
305310315320
alaargtyrtyrargilethrtyrglygluthrglyglyasnserpro
325330335
valglngluphethrvalprolysasnvaltyrthralathrileser
340345350
glyleulysproglyvalasptyrthrilethrvaltyralavalthr
355360365
lysmetargasptyrargproileserileasntyrargthrgluile
370375380
asplysprosergln
385
- 还没有人留言评论。精彩留言会获得点赞!