一种合成多取代α,β不饱和酮的方法与流程
本发明属于化工技术领域,具体涉及一种合成多取代α,β不饱和酮的方法。
背景技术:
多取代α,β不饱和酮,尤其是三取代的α,β不饱和酮广泛的存在于医药中间体,有机材料以及高分子聚合物中,是一种非常重要的合成子。传统的方法合成该类化合物受到底物很大的限制,要求参与反应的两种酮必须是相同的化合物物或一种酮不具有α位的氢原子,这样就不能作为亲电试剂参与反应。否则,往往会得到混合的产物,造成较低的产率和分离的困难。目前合成这类化合物得方法主要是两个酮之前发生adol反应,再经过脱水的到产物。但这种方法要求必须是两种相同的酮发生反应,或者一种酮不具有α位的质子,否则很难控制反应的选择性。
此外,国外的文献也报道了一些合成此类化合物的新方法。例如在(j.am.chem.soc.1981,103,2404-2405)上报道了一种环丙烷开环的方法合成此类化合物的新方法,但该方法需要采用不稳定的三元环作为反应底物,而三元环的合成本身就具有较大的困难。例如在(j.am.chem.soc.2003,125,12576-12583)上报道了一种联烯参与的三组份反应,但该反应也采用高活性的联烯作为底物。为此,有必要发明一种新的合成α,β不饱和酮的方法。
技术实现要素:
为了解决现有技术中存在的上述问题,本发明提供了一种合成多取代α,β不饱和酮的方法,该方法采用了廉价的ni(cod)2作为催化剂,通过过渡金属来活化碳碳键,双键的异构化为该反应提供了动力,合成方法简单,底物稳定廉价,反应受底物限制性少,且底物适用性范围广。
本发明要解决的技术问题通过以下技术方案实现:
一种合成多取代α,β不饱和酮的方法,包括如下步骤:
步骤一:将ni(cod)2、氮杂环类卡宾配体、cs2co3和β,γ不饱和酮加入非极性非质子有机溶剂中,进行混合,制得混合物;
步骤二:将步骤一中的混合物加入干燥后的容器中进行反应,反应温度120~180℃,反应时间10~40小时,反应结束后冷却至室温,制得粗产物。
优选地,上述的合成多取代α,β不饱和酮的方法,还包括步骤三:将步骤二中的粗产物过滤,滤液经真空浓缩后得到滤渣,再用硅胶柱柱层析法对滤渣进行纯化,α,β不饱和酮。
优选地,上述氮杂环类卡宾配体为1,3-二(2,4,6-三甲基苯基)氯化咪唑、1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物、1,3-双(2,6-二异丙基苯基)氯化咪唑翁或1,3-二环己基氯化咪唑。
优选地,上述非极性非质子有机溶剂为二氧六环、甲苯或二甲苯。
优选地,上述ni(cod)2和氮杂环类卡宾配体的摩尔比为1:2。
优选地,上述ni(cod)2的含量为5~15mol%,所述氮杂环类卡宾配体的含量为10~30mol%,所述cs2co3的含量为20~35mol%。
优选地,上述ni(cod)2的含量为10mol%,所述氮杂环类卡宾配体的含量为20mol%,所述cs2co3的含量为30mol%。
优选地,上述反应温度为150℃,反应时间24小时。
优选地,上述的合成多取代α,β不饱和酮的方法,反应式如式(a):
其中,r为:苯基、2-甲基苯基、3-甲基苯基、4-甲基苯基、4-氟代苯基、4-氯代苯基、4-氰基代苯基、4-乙基代苯基、4-甲氧基苯基、苯酚、4-异丙基代苯基、4-叔丁基代苯基、4-苯基代苯基、2-萘基、2-呋喃基、2-噻吩基、苯基五元环状基、2-(2-丙烯基)苯基或苯乙基。
优选地,上述的合成多取代α,β不饱和酮的方法,反应式如式(b):
其中,r1、r2、r3分别选自乙基、苯基、甲基、正己烷基、苯乙基基、(苯并)五元环至八元环基、雄诺龙基、庚烷基或壬烷基中的任一种。
与现有技术相比,本发明的有益效果:
1.本发明的反应体系简单,反应中只需加入催化剂和配体,所采α位含有酰基取代的烯烃化合物类的底物即可,底物稳定易得,制备成本低;
2.本发明的反应体系干净,转化率较高,仅需将体系中的催化剂过滤掉即可,产物易分离;
3.本发明所用方法适用性广泛,大多数常见的官能团的均适用本方法,底物的适用性广泛,按照此方法可制备环状α,β不饱和酮和非环状α,β不饱和酮。
附图说明
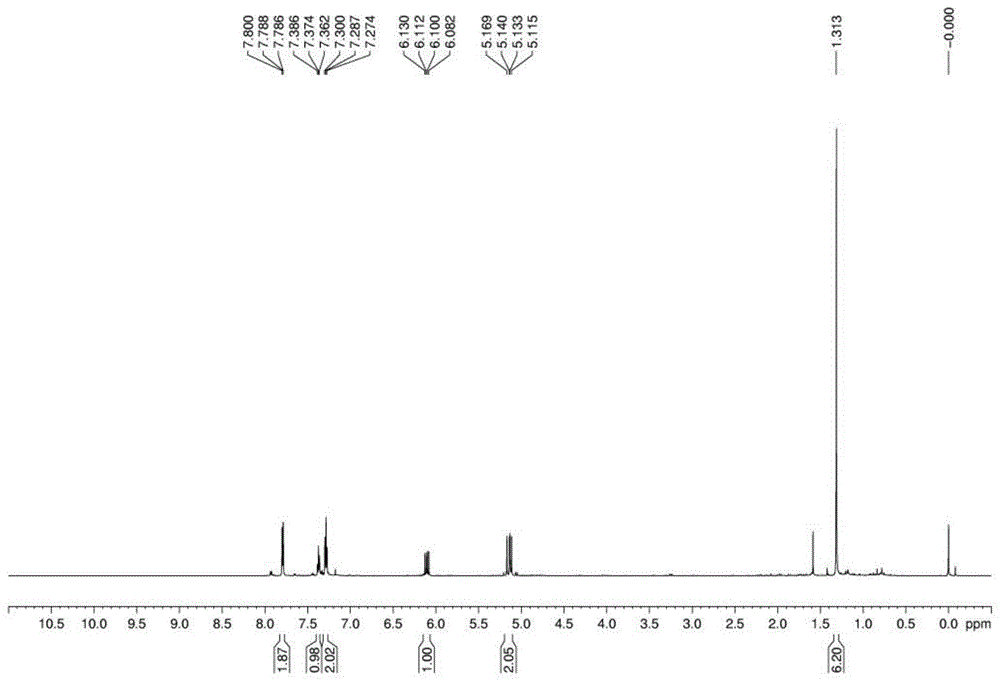
图1是实施例1产物的1hnmr谱图。
图2是实施例1产物的13cnmr谱图。
图3是实施例1产物的x-ray谱图。
图4是实施例1中β,γ不饱和酮的1hnmr谱图。
图5是实施例1中β,γ不饱和酮的13cnmr谱图。
图6是实施例1中β,γ不饱和酮的hrms谱图。
具体实施方式
下面结合具体实施例对本发明做进一步详细的描述,但本发明的实施方式不限于此。
本发明提供了一种合成α,β不饱和酮的方法,包括如下步骤:
步骤一:将5~15mol%的ni(cod)2、10~30mol%的氮杂环类卡宾配体、20~35mol%的cs2co3和β,γ不饱和酮加入非极性非质子有机溶剂中,进行混合,制得混合物;
步骤二:将步骤一中的混合物加入干燥后的容器中进行反应,反应温度120~180℃,反应时间10~40小时,反应结束后冷却至室温,制得粗产物;
步骤三:将步骤二中的粗产物过滤,滤液经真空浓缩后得到滤渣,再用硅胶柱柱层析法对滤渣进行纯化,α,β不饱和酮。
其中,氮杂环类卡宾配体可以为1,3-二(2,4,6-三甲基苯基)氯化咪唑、1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物、1,3-双(2,6-二异丙基苯基)氯化咪唑翁或1,3-二环己基氯化咪唑,氮杂环类卡宾配体可以调节催化剂ni中心的电子效应和立体效应。ni(cod)2和氮杂环类卡宾配体的摩尔比为1:2,1摩尔的ni(cod)2与2摩尔的氮杂环类卡宾配体相配位。考虑到底物与催化剂的溶解性,所以本方法选用的是非极性非质子有机溶剂,本实施例的非极性非质子有机溶剂选用的是二氧六环、甲苯或二甲苯。
本发明的方法根据反应底物结构的不同,通过同一个机理,可合成非环状产物的α,β不饱和酮和环状产物的α,β不饱和酮。
(1)合成非环状产物的α,β不饱和酮的反应式如下:
其中,r为:苯基、2-甲基苯基、3-甲基苯基、4-甲基苯基、4-氟代苯基、4-氯代苯基、4-氰基代苯基、4-乙基代苯基、4-甲氧基苯基、苯酚、4-异丙基代苯基、4-叔丁基代苯基、4-苯基代苯基、2-萘基、2-呋喃基、2-噻吩基、苯基五元环状基、2-(2-丙烯基)苯基或苯乙基。
产物的结构式如下:
(2)合成非环状产物的α,β不饱和酮的反应式如下:
其中,r1、r2、r3分别选自乙基、苯基、甲基、正己烷基、苯乙基基、(苯并)五元环至八元环基、雄诺龙基、庚烷基或壬烷基中的任一种。
产物的结构式如下:
实施例1:
将0.02mmol的ni(cod)2、0.04mmol的1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物(sipr·hcl)、0.06mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的二氧六环中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在150℃,在搅拌条件下反应24小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为76%。
产物通过经nmr、hrms、x-ray检测,产物的1hnmr谱图如图1所示,产物的13cnmr谱图如图2所示,产物的x-ray谱图如图3所示。
参照图1,1hnmr(600mhz,cdcl3):δ7.80–7.79(m,2h),7.39–7.36(m,1h),7.30–7.27(m,2h),6.13–6.08(dd,j=10.8hz,j=18.0hz,1h),5.17–5.12(m,
2h),1.31(s,6h);13cnmr(125mhz,cdcl3):δ204.6,143.7,136.9,131.6,129.2,127.8,113.9,50.0,25.9.hrms(esi)m/zcalculatedforc12h14ona[m+na]+
197.0937,found197.0923。
本实施例中β,γ不饱和酮的合成方法为:
步骤1:在thf/h2o(30.0ml/30.0ml)中加入醛(10.0mmol)和溴化物2(12.0mmol),在0℃下分批加入锌粉(12.0mmol)。反应混合物在室温下搅拌12小时,然后用etoac(3×50ml)进行过滤和提取。结合有机相用盐水洗涤,用na2so4干燥。溶剂在减压下过滤蒸发后,粗品经硅胶柱层析纯化得到醇。
步骤2:将乙醇(10.0mmol)溶于dcm(50.0ml)中,室温下加入pcc(25.0mmol),搅拌3h,通过tlc分析确定乙醇的消耗量。反应混合物经etoac(3×50ml)过滤提取。结合有机相用盐水洗涤,用na2so4干燥。溶剂在减压下过滤蒸发后,粗品经硅胶柱层析纯化得到产物β,γ不饱和酮。产物通过经nmr和hrms检测,产物的1hnmr谱图如图4所示,产物的13cnmr谱图如图5所示,产物的hrms谱图如图6所示。
该合成多取代α,β不饱和酮的方法,采用了廉价的ni(cod)2催化剂作为催化剂,采用了目前最前沿的碳碳键活化的方法,从简单化合物物苯甲醛和烯丙基溴出发,经过三步反应,快速而高效的得到α,β不饱和酮,合成方法简单,底物稳定廉价,底物限制性少。
实施例2:
将0.02mmol的ni(cod)2、0.04mmol的1,3-双(2,6-二异丙基苯基)氯化咪唑翁(ipr)、0.06mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的二氧六环中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在150℃,在搅拌条件下反应24小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为66%。
实施例3:
将0.02mmol的ni(cod)2、0.04mmol的1,3-二(2,4,6-三甲基苯基)氯化咪唑(imes)、0.06mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的二氧六环中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在150℃,在搅拌条件下反应24小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为31%。
实施例4:
将0.02mmol的ni(cod)2、0.04mmol的1,3-二环己基氯化咪唑(icy)、0.06mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的二氧六环中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在150℃,在搅拌条件下反应24小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为46%。
实施例5:
将0.02mmol的ni(cod)2、0.04mmol的1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物(sipr·hcl)、0.06mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的甲苯中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在150℃,在搅拌条件下反应24小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为58%。
实施例6:
将0.02mmol的ni(cod)2、0.04mmol的1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物(sipr·hcl)、0.06mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的二甲苯中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在150℃,在搅拌条件下反应24小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为35%。
实施例7:
将0.02mmol的ni(cod)2、0.04mmol的1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物(sipr·hcl)、0.06mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的甲苯中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在120℃,在搅拌条件下反应24小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为40%。
实施例8:
将0.01mmol的ni(cod)2、0.02mmol的1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物(sipr·hcl)、0.04mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的甲苯中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在180℃,在搅拌条件下反应10小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为50%。
实施例9:
将0.03mmol的ni(cod)2、0.06mmol的1,3-双-(2,6-二异丙基苯基)咪唑鎓氯化物(sipr·hcl)、0.07mmol的cs2co3和0.2mmol的β,γ不饱和酮加入至2ml的甲苯中,进行混合,混合后加入在手套箱中密封干燥后的试管中进行反应,采用油浴加热,温度控制在150℃,在搅拌条件下反应40小时,制得粗混合物。将粗混合物自然冷却到室温,再通过硅胶垫过滤,滤液经真空浓缩后得到滤渣,用硅胶柱柱层析法使用乙酸乙酯与石油醚进行纯化,得到α,β不饱和酮。产率为62%。
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!