六元脂环酰亚胺类衍生物单体及其制备、应用、固化邻苯二甲腈树脂的方法与流程
本发明涉及材料加工技术领域,尤其涉及六元脂环酰亚胺类衍生物单体及其制备、应用、固化邻苯二甲腈树脂的方法。
背景技术:
邻苯二甲腈树脂(pn树脂)是一类非常重要的高强耐高温聚合物材料,具有高玻璃化转变温度(tg),优异的热氧稳定性,突出的机械和物理化学性能,卓越的阻燃性能,在航空航天、舰船与海洋工程、交通运输、微电子封装等高端装备及国防军工领域展现出了广阔的应用前景。然而,分子结构的刚性、低反应活性等因素使得纯邻苯二甲腈单体的固化非常缓慢,需要在280℃热处理近百小时才能观察到明显的凝胶,这大大限制了该树脂体系的加工应用。
目前邻苯二甲腈树脂的固化体系主要有两大类,一类是双组分或多组分催化固化体系,另一类是自催化邻苯二甲腈体系。但这些固化方法仍未能有效提高固化效率,解决加工难题。一方面,酚、芳香族伯胺、有机强酸、酸酐、有机酸/铵盐及金属盐等固化剂或添加剂可以缩短固化时间,加速邻苯二甲腈树脂的固化。但是,过渡金属、过渡金属盐等在体系中难以形成分子水平的分散和高温下金属易氧化的特征极大地影响了材料的最终性能;且这类双组分或多组分催化固化剂大多为小分子物质,耐热性不够,高温下易挥发造成配方工艺的波动影响固化效果。另一方面,一类自身含有氨基或羟基等“活性氢”结构的自催化邻苯二甲腈树脂体系中,氰基的固化也并不完全,固化速率不高,后固化仍需在较高温度下进行长时间热处理。且固化后的这类树脂由于高交联度、高刚性而具有脆性。同时,其加工性也因为邻苯二甲腈化合物的高熔点或软化点和较窄的加工温度窗而受到限制。相关机理以亲核加成固化历程为主。而利用苯乙炔基固化放热量大的特性虽然可以催化邻苯二甲腈固化,但苯乙炔单体的成本较高,且固化温度较高,在350℃左右,对固化设备要求严格。总之,不管是通过配方与固化工艺优化,还是提高性能和降低成本方面,pn树脂的固化加工性较差的问题仍未从根本上有效解决。因此,开发新型高效固化剂成为解决邻苯二甲腈树脂固化难题的一种重要途径。
技术实现要素:
本发明为了解决上述技术问题提供了一类可与双邻苯二甲腈发生协同固化作用的六元脂环酰亚胺类衍生物单体。
本发明公开的六元脂环酰亚胺类衍生物单体,包括端基六元脂环酰亚胺单体和分子内六元脂环酰亚胺单体,其化学通式分别为:
其中,r1选自:
r2为:
其中,ar1选自:
中的任一种;
其中,ar2选自:
本发明还提供了上述的六元脂环酰亚胺类衍生物单体的制备方法,该制备简单易行。
本申请文件公开的六元脂环酰亚胺类衍生物单体的制备方法,采用六元脂环单酸酐或六元脂环二酸酐与二胺类或单胺类化合物反应脱水制备。
其具体包括以下步骤:
s1)按2:1的摩尔比将六元脂环单酸酐、二胺类化合物分别加入溶剂中,室温搅拌反应1~2h后加入三乙胺,得到反应液;
s2)将反应液油浴升温至120~160℃,并继续反应2~5h,直至反应完全;
s3)反应完全后降温,然后将反应产物用水沉淀后,再依次水洗、抽滤、干燥,得到六元脂环酰亚胺类衍生物单体;
其溶剂为dmac或dmf、nmp中任一种。
上述六元脂环酰亚胺类衍生物单体还可以采用以下方法制备:
s1)按2:1的摩尔比将六元脂环单酸酐与二胺类化合物,室温搅拌反应1~2h后加入甲苯或者二甲苯,得到反应液;
s2)将反应液油浴升温回流,共沸脱水3~5h待无明显水分分出后,再分出甲苯或二甲苯;
s3)甲苯或二甲苯分出后,降温,然后将反应产物转移至乙醇中沉淀,再依次水洗、抽滤、干燥,得到六元脂环酰亚胺类衍生物单体;
其溶剂为dmac或dmf。
上述六元脂环酰亚胺类衍生物单体还可以采用以下方法制备:
s1)按1:2的摩尔比将六元脂环二酸酐与单胺类化合物分别加入溶剂中,室温搅拌反应1~2h后加入甲苯或者二甲苯,得到反应液;
s2)将反应液油浴升温回流,共沸脱水3~5h待无明显水分分出后,再分出甲苯或二甲苯;
s3)甲苯或二甲苯分出后,降温,然后将反应产物转移至乙醇中沉淀,再依次水洗、抽滤、干燥,得到六元脂环酰亚胺类衍生物单体;
其溶剂为dmac或dmf。
本发明还提供了六元脂环酰亚胺类衍生物单体在邻苯二甲腈树脂固化上的应用。
本申请文件公开的六元脂环酰亚胺类衍生物单体在邻苯二甲腈树脂固化上的应用是采用六元脂环酰亚胺类衍生物单体与邻苯二甲腈树脂的原料的发生协同固化作用,实现邻苯二甲腈树脂的低温高效固化。
所述邻苯二甲腈树脂的原料包括双邻苯二甲腈单体和/或双邻苯二甲腈低聚物。
其中,双邻苯二甲腈低聚物的分子结构通式为:
其中,n为不小于1的自然数;
其中,r选自:
其中,r’选自:
其中,r”选自:
本发明还提供六元脂环酰亚胺类衍生物单体固化邻苯二甲腈树脂的方法。
六元脂环酰亚胺类衍生物单体固化邻苯二甲腈树脂的方法,包括将六元脂环酰亚胺类衍生物单体与双邻苯二甲腈单体或双邻苯二甲腈低聚物共混,然后在惰性气氛中将得到的共混物进行升温或等温热处理,再在230~320℃条件下等温固化3~9h,得到固化完全的邻苯二甲腈树脂。
进一步地,所述六元脂环酰亚胺类衍生物单体与双邻苯二甲腈单体或双邻苯二甲腈低聚物的摩尔比为1:4~1:1。
进一步地,所述六元脂环酰亚胺类衍生物单体与双邻苯二甲腈单体或双邻苯二甲腈低聚物的共混物通过以下步骤得到:按摩尔比取六元脂环酰亚胺类衍生物单体与双邻苯二甲腈单体或双邻苯二甲腈低聚物,在低沸点溶剂中进行共混,然后减压蒸馏除去低沸点溶剂后,再研磨成粉,得到共混物。
附图说明
下述描述中的附图仅仅是本申请的一些实施例,对于本领域技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
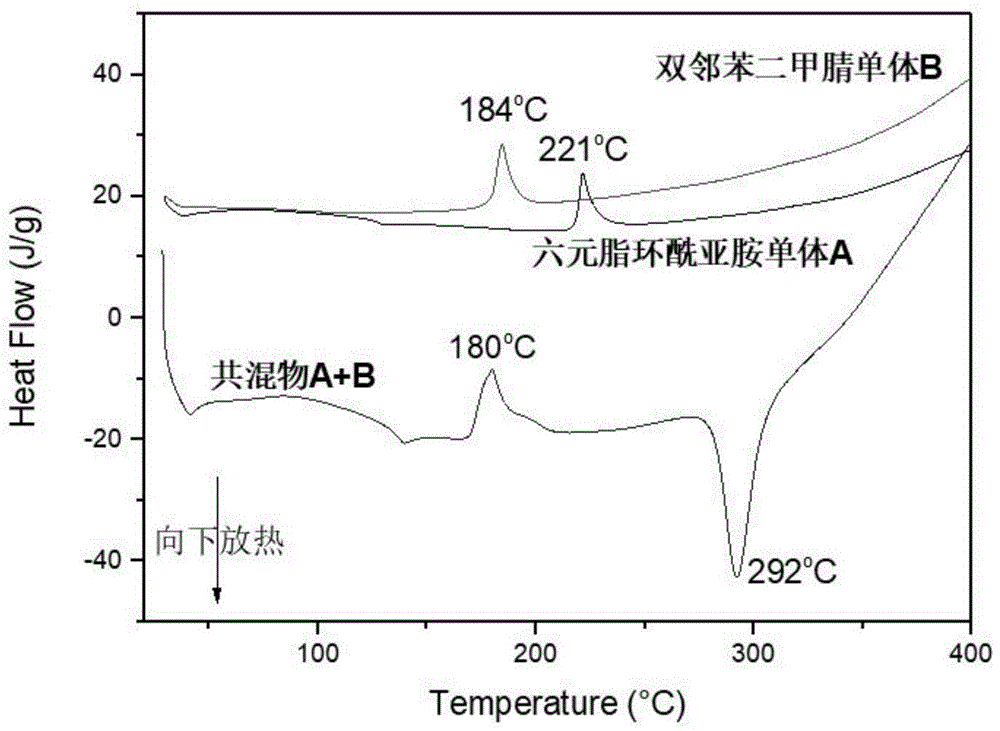
图1本发明的一些实施例中制备的六元脂环酰亚胺单体a和双邻苯二甲腈单体b及两者的共混物的dsc图;
图2本发明的一些实施例中制备的六元脂环酰亚胺单体a和双邻苯二甲腈单体b及两者的共混物的tga图。
具体实施方式
为了使本技术领域人员更好地理解本发明方案,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
下述实施例中,六元脂环酰亚胺单体可通过以下方法一制备:
在dmac(n,n-二甲基乙酰胺)或dmf(n,n-二甲基甲酰胺)或nmp(n-甲基吡咯烷酮)溶剂中,按照1:2的摩尔比分别加入二胺类化合物和六元脂环单酸酐,室温搅拌反应1-2h后加入三乙胺,油浴升温至120-160℃反应2-5h脱水闭环,通过薄层层析色谱(tlc)监控反应进程,待原料反应完全。降至室温,反应产物用水沉淀后,再水洗、抽滤,干燥,得到六元脂环酰亚胺单体。
六元脂环酰亚胺单体可通过以下方法二制备:
在dmac或nmp溶剂中,按照1:2的摩尔比分别加入二胺类化合物和六元脂环单酸酐,室温搅拌反应1-2h后加入甲苯或二甲苯,油浴升温至98~105℃回流,共沸脱水3-5h待无明显水分出后,再升温至110~130℃,分出甲苯或二甲苯,降至室温,反应产物转移至乙醇中沉淀后,再用大量水洗、抽滤,干燥,得到六元脂环酰亚胺单体。
六元脂环酰亚胺单体可通过以下方法三制备:
在dmac或nmp溶剂中,按照2:1的摩尔比分别加入单胺类化合物和六元脂环二酸酐,室温搅拌反应1-2h后加入甲苯或二甲苯,油浴升温至98~105℃回流,共沸脱水3-5h待无明显水分出后,再升温至110~130℃,分出甲苯或二甲苯,降至室温,反应产物转移至乙醇中沉淀后,再用大量水洗、抽滤,干燥,得到六元脂环酰亚胺单体。
下述实施例中的双邻苯二甲腈单体或其低聚物可通过以下方法制备:
在dmso或dmf等溶剂中,加入二酚或间苯三酚或端羟基聚硅氧烷、3硝基邻苯二甲腈或4-硝基邻苯二甲腈以及研磨较细的碳酸钾粉末,氮气保护下室温或升至一定温度如40℃等温反应12-24h左右,反应液用水沉淀,大量水洗、抽滤,干燥,得到双邻苯二甲腈单体或其低聚物。
六元脂环酰亚胺单体与双邻苯二甲腈单体或其低聚物的共混物采用如下方法制备:按照1:4~1:1的摩尔比称取六元脂环酰亚胺单体与邻苯二甲腈单体或其低聚物,并加入低沸点溶剂中,进行搅拌混合均匀,然后通过减压蒸馏除去低沸点溶剂后,研磨成粉,得到共混物。
可选地,低沸点溶剂为乙醇或丙酮。
对六元脂环酰亚胺单体、邻苯二甲腈单体以及六元脂环酰亚胺单体与邻苯二甲腈单体的共混物进行热分析,其中热分析得到的tga曲线和dsc曲线,如图1、图2所示,六元脂环酰亚胺单体与邻苯二甲腈单体的共混物在400℃以内出现有明显的放热峰,而六元脂环酰亚胺单体a、邻苯二甲腈单体b则没有出现明显的放热峰;而对六元脂环酰亚胺单体、邻苯二甲腈单体或其低聚物在700℃的残炭率不超过20%;共混物在在700℃的残炭率则超过60%。
由此可见,六元脂环酰亚胺单体a与邻苯二甲腈单体b的共混物发生了协同固化作用。
实施例1
以六元脂环酰亚胺单体为1,2,3,6-四氢苯酐型封端酰亚胺单体、双邻苯二甲腈单体为羟基二苯醚型双邻苯二甲腈单体为例:
其化学结构式分别为:
将1,2,3,6-四氢苯酐型封端酰亚胺单体与二羟基二苯醚型双邻苯二甲腈单体按3:7摩尔比例按上述共混方法进行物理共混后,研磨成细粉,并对其共混物、1,2,3,6-四氢苯酐型封端酰亚胺单体、羟基二苯醚型双邻苯二甲腈单体进行热分析,并对其热分析后的样品进行红外分析。其中热分析得到的tga曲线和dsc曲线中:700℃时1,2,3,6-四氢苯酐型封端酰亚胺单体与二羟基二苯醚型双邻苯二甲腈单体的残炭率分别仅为13.1%和11.1%,但其共混物的残炭率达到了56%;在350℃范围内,上述两种单体均无明显放热峰出现,但其共混物在322℃附近则出现了明显的放热峰,dsc测试完毕后共混物样品的红外测试结果显示,2232cm-1处c≡n几乎反应完全。将共混物215℃附近熔融后再在230℃等温处理1h,c≡n转化率达到83%。
实施例2
以六元脂环酰亚胺单体为3,4,5,6-四氢苯酐型封端酰亚胺单体、双邻苯二甲腈单体为双酚芴型双邻苯二甲腈单体为例:
其化学结构式分别为:
将3,4,5,6-四氢苯酐型封端酰亚胺单体与双酚芴型双邻苯二甲腈单体按5:5摩尔比例进行物理共混后,研磨成细粉,对其共混物、3,4,5,6-四氢苯酐型封端酰亚胺单体、双酚芴型双邻苯二甲腈单体进行热分析,并对其热分析后的样品进行红外分析。其中热分析得到的tga曲线和dsc曲线中:700℃时3,4,5,6-四氢苯酐型封端酰亚胺单体与双酚芴型双邻苯二甲腈单体的残炭率分别为4.5%和54.4%,其共混物的残炭率达到了75%;在350℃范围内,上述两种单体均无明显放热峰出现,但其共混物在340℃附近则出现了明显的放热峰,dsc测试完毕后的共混物样品的红外测试结果显示,2233cm-1处c≡n几乎反应完全。将共混物在260℃附近熔融后再等温处理1h,c≡n转化率达到79%。
实施例3
以六元脂环酰亚胺单体为4-甲基六氢苯酐型封端酰亚胺单体和六氢苯酐型封端酰亚胺单体、双邻苯二甲腈单体为间苯三酚型双邻苯二甲腈单体为例,
其化学结构式分别为:
将4-甲基六氢苯酐型封端酰亚胺单体、六氢苯酐型封端酰亚胺单体以及间苯三酚型双邻苯二甲腈单体按4:1:5摩尔比例共混后,研磨成细粉,得到共混物,并对其共混物、4-甲基六氢苯酐型封端酰亚胺单体、六氢苯酐型封端酰亚胺单体、间苯三酚型双邻苯二甲腈单体进行热分析,并对其热分析后的样品进行红外分析。其中热分析得到的tga曲线和dsc曲线中:700℃时4-甲基六氢苯酐型封端酰亚胺单体、六氢苯酐型封端酰亚胺单体与间苯三酚型双邻苯二甲腈单体的残炭率分别为15.2%、0%和13.7%,其共混物的残炭率达到了68%,在350℃范围内,上述三种单体均无明显放热峰出现,但其共混物在338℃附近则出现了明显的放热峰,dsc测试完毕后的共混物样品的红外结果显示,2233cm-1处c≡n吸收峰强度明显减弱。进行氮气氛下对共混物进行原位升温红外测试。测试温度为室温至350℃,测试时间共计约为20min。原位红外测试结果显示,150℃前,2233cm-1处c≡n红外吸收峰强度无明显变化,255℃后,2950cm-1附近的甲基以及脂环特征峰和2233cm-1处c≡n峰强度开始变弱,两类官能团的转化率开始升高。295℃时,2233cm-1处c≡n转化率为41%;330℃时c≡n转化率为65%;到350℃时c≡n转化率则达到了80%。
实施例4
以六元脂环酰亚胺单体为4-甲基四氢苯酐型封端酰亚胺单体、双邻苯二甲腈单体为醚酐酰亚胺型双邻苯二甲腈单体为例,
其化学结构式分别为:
将4-甲基四氢苯酐型封端酰亚胺单体与醚酐酰亚胺型双邻苯二甲腈单体按5:5的摩尔比进行共混后,研磨成细粉,得到共混物,并对其共混物、4-甲基四氢苯酐型封端酰亚胺单体、醚酐酰亚胺型双邻苯二甲腈单体进行热分析,并对其热分析后的样品进行红外分析。其中热分析得到的tga曲线和dsc曲线中:700℃时4-甲基四氢苯酐型封端酰亚胺单体与醚酐酰亚胺型双邻苯二甲腈单体的残炭率分别为9.4%和38.1%,其共混物的残炭率达到了67%;在350℃范围内,上述两种单体均无明显放热峰出现,但其共混物在328℃附近则出现了明显的放热峰,dsc测试完毕后的样品红外结果显示,2234cm-1处c≡n几乎反应完全。将共混物在270℃附近熔融后再等温处理1h,c≡n转化率达到63%。
实施例5
以六元脂环酰亚胺单体为环己烷四酸二酐型酰亚胺单体、双邻苯二甲腈单体为聚芳醚腈型双邻苯二甲腈和聚苯基膦酰氧醚型双邻苯二甲腈为例,
其化学结构式分别为:
将环己烷四酸二酐型酰亚胺单体、聚芳醚腈型双邻苯二甲腈以及聚苯基膦酰氧醚型双邻苯二甲腈按3:1:1质量比例共混后,研磨成细粉得到共混物,并对其共混物、环己烷四酸二酐型酰亚胺单体、聚芳醚腈型双邻苯二甲腈、聚苯基膦酰氧醚型双邻苯二甲腈进行热分析,并对其热分析后的样品进行红外分析。其中热分析得到的tga曲线和dsc曲线中:700℃时环己烷四酸二酐型酰亚胺单体、聚芳醚腈型双邻苯二甲腈与聚苯基膦酰氧醚型双邻苯二甲腈的残炭率分别为46.9%、33.9%和31%,其共混物的残炭率达到了72%,在350℃范围内,上述三种单体和低聚物均无明显放热峰出现,其共混物在335℃附近则出现了明显的放热峰,dsc测试完毕后的共混物的红外结果显示,2234cm-1处c≡n吸收峰强度明显减弱。进行氮气氛下对共混物进行原位升温红外测试。测试温度为室温至350℃,测试时间共计约为20min。原位红外测试结果显示,170℃前,2234cm-1处c≡n红外吸收峰强度无明显变化,275℃后,2950cm-1附近的亚甲基以及脂环特征峰和2234cm-1处c≡n峰强度开始变弱,两类官能团的转化率开始升高,350℃时c≡n转化率则达到了69%。
综上,六元脂环酰亚胺单体与双邻苯二甲腈单体和/或其低聚物共混后,进行加热固化,产生协同固化作用,使双邻苯二甲腈单体和/或低聚物在较低温度下即可达到较高效的固化,实现双邻苯二甲腈单体和/或低聚物的低温高效固化,可应用于邻苯二甲腈树脂的固化工艺中。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!