苯酰基修饰的聚集诱导发光型苯胺齐聚物及其制备方法与流程
本发明属于光致发光材料领域,尤其涉及一种苯酰基修饰的聚集诱导发光型苯胺齐聚物及其制备方法。
背景技术:
传统的有机发光材料在聚集过程中会由于π-π堆积作用发生非辐射能量转移,导致自身发光在很大程度上淬灭。一些特殊的“螺旋桨”结构的芳香化合物,在溶解状态下由于“螺旋桨”的转动消耗激发态分子能量,因此荧光十分微弱,而在聚集状态下由于扭曲的分子构象,荧光分子很难形成较好的平面性,有效地避免了π-π堆积,极大地降低了非辐射能量消耗,荧光强度表现出明显增强,即聚集诱导发光(aggregation-inducedemission,aie)。具有aie性能的新材料从根本上克服了聚集导致荧光淬灭(aggregationcausedquenching,acq)的难题,因此成为新的前沿研究热点。这些化合物分子产生aie现象的机理通常认为是分子内旋转受限导致的。
基于aie机理,许多aie化合物被设计合成出来。常见的aie化合物有噻咯、芳基取代的环丁烯、乙烯(如四苯乙烯tpe)和丁二烯以及吡喃衍生物等。然而,这些分子存在着一些不容忽视的问题,例如,多苯基噻咯的制备过程繁琐,分离困难且对碱不稳定。此外,与大量研究的传统acq体系相比,aie体系仍然处于探索的阶段,材料种类还很有限,因此设计开发多种易于合成的新结构、新种类发光性能优良的aie化合物有很好的学术价值和应用价值。
苯胺齐聚物类化合物价格低廉,合成简单,环境稳定性好,具有特殊的链结构和优良的物理性能,在光电功能材料和器件领域具有广阔的应用前景。然而,大多数苯胺齐聚物都受到acq效应的影响,这是因为存在着不同的分子间相互作用,包括π-π堆积、疏水作用和氢键作用,这极大地影响了它们在发光领域的应用。
技术实现要素:
本发明制备出一类含苯酰基苯胺齐聚物衍生物,利用苯胺齐聚物和酰氯同系物为初始反应物,通过在苯胺齐聚物上连接苯酰胺,从而得到新型含苯酰基苯胺齐聚物衍生物,解决了传统苯胺齐聚物聚集诱导淬灭的问题,使其具有聚集诱导发光的特性。通过本发明可以简单、高效地获得一系列具有aie性能的含苯酰基苯胺齐聚物衍生物。另外,通过侧基的控制,可以实现不同苯酰基修饰的苯胺齐聚物衍生物的可调谐发光。
本发明的目的之一在于提供一种苯酰基修饰的聚集诱导发光型苯胺齐聚物,包括如下化学式i:
其中,r1和r2分别独立地为以下官能团(从左至右分别为苯甲酰基、苯乙酰基和二苯乙酰基):
当n=1时,当r1和r2均为苯甲酰基时,对应的苯胺二聚体衍生物简写为c2-a2;当r1和r2均为苯乙酰基时,对应的苯胺二聚体衍生物简写为c2-b2;当r1和r2均为二苯乙酰基时,对应的苯胺二聚体衍生物简写为c2-c2。
优选地,苯胺二聚体衍生物c2-a2为具备aie性能的白色固体,在365nm紫外灯下呈绿色;苯胺二聚体衍生物c2-b2为具备aie性能的白色固体c2-b2,在365nm紫外灯下呈青色;苯胺二聚体衍生物c2-c2为具备aie性能的淡黄色固体c2-c2,在365nm紫外灯下呈蓝色。
本发明还提供了一种上述苯酰基修饰的聚集诱导发光型苯胺齐聚物的制备方法,合成路线如下:
优选地,苯胺二聚体衍生物c2-a2的制备步骤包括:
步骤101:将n,n’-二苯基-1,4-对苯二胺放置于三口烧瓶中,分别加入吡啶做缚酸剂、三氯甲烷做溶剂充分溶解,得到溶液,n,n’-二苯基-1,4-对苯二胺、吡啶和三氯甲烷的摩尔比为3.8:1:250;
步骤102:将苯甲酰氯溶解于三氯甲烷中稀释,苯甲酰氯与三氯甲烷的体积比为2:5,在冰水浴中缓慢滴加于步骤101中所得溶液并均匀搅拌,滴加完毕后将溶液移到室温下反应4个小时,反应结束后得到棕红色溶液;
步骤103:对步骤102中得到的棕红色溶液过滤取滤液,将滤液用三氯甲烷稀释,用盐酸水溶液洗涤除去过量吡啶及生成的吡啶盐酸盐,后用水复洗涤至ph为7左右;分液取有机层,加入适量无水硫酸钠除水,过滤取滤液,旋蒸得固体产物;接着采用沉淀法,利用二氯甲烷和乙酸乙酯溶解所得固体产物,其中,二氯甲烷与乙酸乙酯的体积比为2:50,并使固体产物在过量乙酸乙酯溶液中缓慢沉淀析出,制备得到苯胺二聚体衍生物c2-a2。
优选地,制备得到的苯胺二聚体衍生物c2-a2的产率约75%。
优选地,苯胺二聚体衍生物c2-b2的制备步骤包括:
步骤201:将n,n’-二苯基-1,4-对苯二胺放置于三口烧瓶中,分别加入吡啶做缚酸剂、三氯甲烷做溶剂充分溶解,得到溶液,n,n’-二苯基-1,4-对苯二胺、吡啶和三氯甲烷的摩尔比为3.8:1:250;
步骤202:将苯乙酰氯溶解于三氯甲烷中稀释,苯乙酰氯与三氯甲烷的体积比为3:5,在冰水浴中缓慢滴加于步骤201中所得溶液并均匀搅拌,滴加完毕后将溶液移到室温下反应5个小时;
步骤203:对步骤202中得到的溶液过滤取滤液,用过量三氯甲烷与盐酸水溶液萃取,后用水溶液反复洗涤至ph为7左右,除去过量吡啶及吡啶盐酸盐;分液取有机层,加入适量无水硫酸钠除水,过滤取滤液,旋蒸得固体产物;接着采用沉淀法,利用二氯甲烷和乙酸乙酯溶解所得固体产物,其中,二氯甲烷与乙酸乙酯的体积比为2:50,并使固体产物在过量乙酸乙酯溶液中缓慢沉淀析出,得到苯胺二聚体衍生物c2-b2。
优选地,制备得到的苯胺二聚体衍生物c2-b2的产率约80%。
优选地,苯胺二聚体衍生物c2-c2的制备步骤包括:
步骤301:将n,n’-二苯基-1,4-对苯二胺放置于三口烧瓶中,分别加入吡啶做缚酸剂、二氯甲烷做溶剂充分溶解,得到溶液,n,n’-二苯基-1,4-对苯二胺、吡啶和二氯甲烷的摩尔比为3.8:1:250;
步骤302:将二苯基乙酰氯溶解于二氯甲烷中稀释,二苯基乙酰氯与二氯甲烷的体积比为1:3,在冰水浴中缓慢滴加于步骤301中所得溶液并均匀搅拌,滴加完毕后将溶液移到室温下反应4个小时;
步骤303:对步骤302中得到的溶液过滤取滤液,用过量二氯甲烷与盐酸水溶液萃取,后用水溶液反复洗涤至ph为7左右,除去过量吡啶及吡啶盐酸盐;分液取有机层,加入适量无水硫酸钠除水,过滤取滤液,旋蒸得固体产物;之后用柱层析法,展开剂石油醚与乙酸乙酯的体积比为4:1,分离提纯,得到苯胺二聚体衍生物c2-c2。
优选地,制备得到的苯胺二聚体衍生物c2-c2的产率约71%。
本发明的有益效果:
1)本发明提供的苯胺二聚体衍生物,由于苯甲酰氯和苯乙酰氯立体基团的引入,使苯胺二聚体衍生物具有“螺旋桨”结构,减少了高浓度或者固体状态下堆积结构的形成,从而解决苯胺二聚体衍生物聚集诱导淬灭的问题,使其具有aie特性;
2)本发明提供的合成路线简单,通过简单的官能团反应即可得到一系列具有aie性能的苯胺二聚体衍生物;
3)本发明提供的反应条件温和,操作简单,原料廉价易得,产率高;
4)本发明提供的合成路线只需要一步就可以得到目标产物,反应流程简化,易于控制,降低了多步合成中的错误几率;
5)本发明提供的制备方法的整个制备过程主要在常温常压下进行,所使用试剂、溶剂均常见低毒,操作步骤简单,所需设备少,综合成本较低。
附图说明
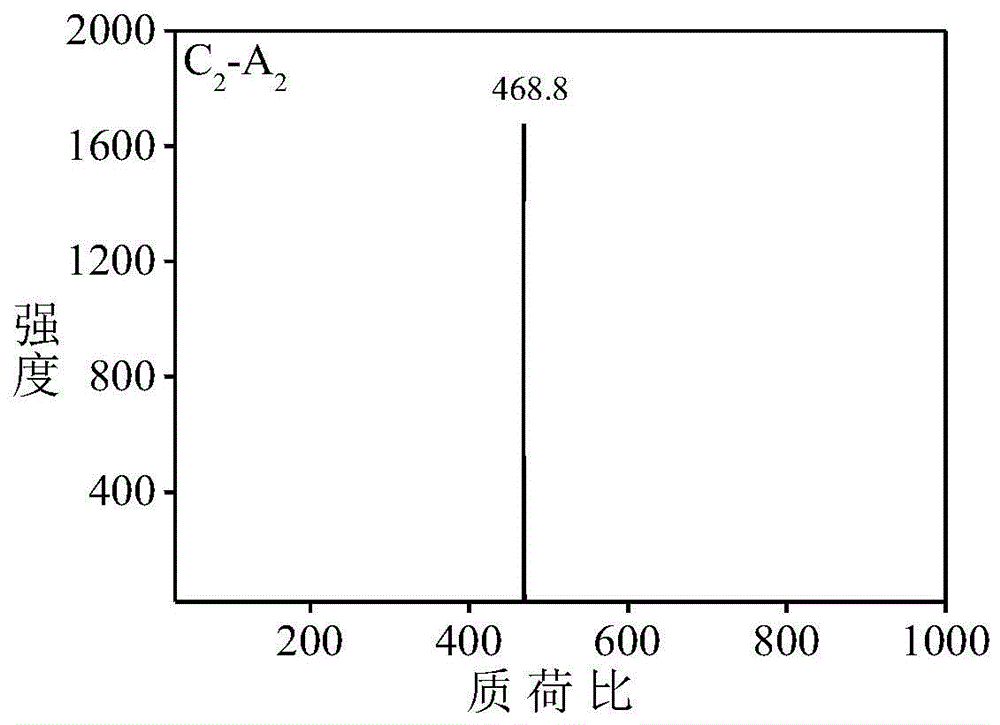
图1是本发明实施例1得到的化合物c2-a2的质谱图;
图2是本发明实施例2得到的化合物c2-b2的质谱图;
图3是本发明实施例3得到的化合物c2-c2的质谱图;
图4(a)-(c)分别示出了本发明化合物c2-a2、c2-b2和c2-c2在混合溶液中的荧光谱图(混合溶液体系分别为:a-正己烷/三氯甲烷体系;b-正己烷/三氯甲烷体系;c-水/dmf体系)
具体实施方式
除另有规定外,在此使用的全部技术和科学术语都具有和本发明所属的领域中技术人员通常所理解的含义。
在本发明全文中,在描述某些方法包括或具有某些特定步骤时,其意味着本发明的方法可以基本由或是由这些引用的步骤组成。
除另有定义外,术语“包括”、“具有”通常应该被理解成是开放式且非限制式的。
除另有说明外,本发明中所有的表示数量,百分比或比例的数值,或其他在本发明说明书或权利要求书中使用的其他数值,都应该为理解为是大约的。除另有说明外,在说明书中出现的数值是估计的,并且取决于试图获得的性质。至少,每个数值参数至少应被解释为根据记载的重要数值通过应用普通的四舍五入获得。
另外除特别定义外,在数值之前用到术语“约”或“大约”时,本教导包括该值本身。除特别定义外,在此使用的术语“约”或“大约”包括数值的正负10%的差值。
下面结合附图和实施例来详细说明本发明,应该理解,以下详细说明旨在便于对本发明的理解,而对其不起任何限定作用。
本发明提供的一系列苯酰基修饰的聚集诱导发光型苯胺齐聚物,包括如下化学式i:
其中,r1和r2分别独立地为以下官能团(从左至右分别为苯甲酰基、苯乙酰基和二苯乙酰基):
其中,n=1时,苯胺二聚体衍生物c2-a2的结构特征为:苯胺二聚体为骨架,苯甲酰基为取代基;苯胺二聚体衍生物c2-b2的结构特征为:苯胺二聚体为骨架,苯乙酰基为取代基;苯胺二聚体衍生物c2-c2的结构特征为:苯胺二聚体为骨架,二苯乙酰基为取代基。
实施例1
苯胺二聚体衍生物c2-a2的制备
苯胺二聚体衍生物c2-a2的合成路线如下所示:
具体包括如下步骤:
步骤101:称取1gn,n’-二苯基-1,4-对苯二胺放置于100ml三口烧瓶中,分别加入3ml吡啶做缚酸剂、20ml三氯甲烷做溶剂充分溶解,得到溶液;
步骤102:量取6ml苯甲酰氯溶解于15ml三氯甲烷中稀释,在冰水浴中缓慢滴加于烧瓶并均匀搅拌,滴加完毕后将装置移到室温下反应4个小时,反应过程中不断点板监测反应进程。反应结束后得棕红色溶液。
步骤103:反应结束之后,过滤取滤液,将滤液用三氯甲烷稀释,用盐酸水溶液洗涤除去过量吡啶及生成的吡啶盐酸盐,后用水溶液反复洗涤至ph为7左右;分液取有机层,加入适量无水硫酸钠除水,过滤取滤液,旋蒸得固体;接着采用沉淀法,利用二氯甲烷和乙酸乙酯溶解所得固体产物,其中,二氯甲烷与乙酸乙酯的体积比为2:50,并使产物在过量乙酸乙酯溶液中缓慢沉淀析出。最终,得到产率约75%的白色固体c2-a2。对于产物c2-a2选取三氯甲烷为良溶剂,正己烷为不良溶剂。图4(a)可以看出在纯三氯甲烷溶液中,c2-a2发光及其微弱,随着体系中不良溶剂(正己烷)比例的增加,体系的荧光发射强度总体呈不断增强趋势。通过荧光发射图谱,可以观察到当体系中正己烷的含量在0~50%之间,体系的荧光强度变化不大,当正己烷含量达到60%时荧光强度显著增加。对应于荧光图谱即表现为,图谱峰位在565nm处,随不良溶剂体积分数的增大,荧光峰不断增强。正己烷含量90%时体系的荧光强度约为纯三氯甲烷中体系荧光发光强度的8.5倍。以上结果证明产物c2-a2是具有aie性能的化合物。
实施例2
苯胺二聚体衍生物c2-b2的制备
苯胺二聚体衍生物c2-b2的合成路线如下所示:
具体包括如下步骤:
步骤201:称取1gn,n’-二苯基-1,4-对苯二胺放置于100ml三口烧瓶中,分别加入3ml吡啶做缚酸剂、25ml三氯甲烷做溶剂充分溶解,得到溶液;
步骤202:量取6ml苯乙酰氯溶解于10ml三氯甲烷中稀释,在冰水浴中缓慢滴加于烧瓶并均匀搅拌,滴完之后将装置移到室温下反应5个小时,反应过程中不断点板监测反应进程;
步骤203:反应结束之后,过滤取滤液,用过量三氯甲烷与盐酸水溶液萃取,后用水溶液反复洗涤至ph为7左右,除去过量吡啶及吡啶盐酸盐;分液取有机层,加入适量无水硫酸钠除水,过滤取滤液,旋蒸得固体;接着采用沉淀法,利用二氯甲烷和乙酸乙酯溶解所得固体产物,其中,二氯甲烷与乙酸乙酯的体积比为2:50,并使产物在过量乙酸乙酯溶液中缓慢沉淀析出。得产率约80%的白色固体c2-b2。对于产物c2-b2选取三氯甲烷为良溶剂,正己烷为不良溶剂。图4(b)可以看出在纯三氯甲烷溶液中,c2-b2发光及其微弱,随着体系中不良溶剂(正己烷)比例的增加,体系的荧光发射强度总体呈不断增强趋势,可以观察到当体系中正己烷的含量在0~40%之间,体系的荧光强度较弱且变化不大,当正己烷含量达到50%时荧光强度显著增加。对应于荧光图谱即表现为,图谱峰位在480nm处,随不良溶剂体积分数的增大,荧光峰不断增强。正己烷含量为90%时c2-b2溶液体系的荧光强度约为纯三氯甲烷中c2-b2溶液体系荧光发光强度的8倍。以上结果证明c2-b2是具有aie性能的化合物。
实施例3
苯胺二聚体衍生物c2-c2的制备
苯胺二聚体衍生物c2-c2的合成路线如下所示:
具体包括如下步骤:
步骤301:称取1gn,n’-二苯基-1,4-对苯二胺放置于100ml三口烧瓶中,分别加入3ml吡啶做缚酸剂、25ml二氯甲烷做溶剂充分溶解,得到溶液;
步骤302:量取2.6ml的二苯基乙酰氯溶解于10ml二氯甲烷中稀释,在冰水浴中缓慢滴加于烧瓶并均匀搅拌,滴完之后将装置移到室温下反应4个小时,反应过程中不断点板监测反应进程;
步骤303:反应结束之后,过滤取滤液,用过量二氯甲烷与盐酸水溶液萃取,后用水溶液反复洗涤至ph为7左右,除去过量吡啶及吡啶盐酸盐。分液取有机层,加入适量无水硫酸钠除水,过滤取滤液,旋蒸得固体;用柱层析法,展开剂石油醚与乙酸乙酯的体积比为4:1,分离提纯,得产率约71%的淡黄色固体c2-c2。对于产物c2-c2选取dmf为良溶剂,h2o为不良溶剂。图4(c)可以看出在纯dmf溶液中,c2-c2就有微弱的蓝色荧光发射,随着体系中不良溶剂比例的增加,体系的荧光发射强度总体呈不断增强趋势,可以观察到当体系中正己烷的含量在0~50%之间,体系的荧光强度相对较弱变化不大,当水含量达到60%时荧光强度显著增加。对应于荧光图谱即表现为,图谱峰位在440nm处,随不良溶剂体积分数的增大,荧光峰不断增强。不良溶剂水含量90%时c2-c2溶液体系的荧光强度约为纯dmf中c2-c2溶液体系荧光发光强度的10倍。以上结果表明c2-c2是具有aie性能的化合物。
本发明基于aie机理,在苯胺齐聚物上引入含苯酰基立体基团,减少了堆积结构的形成,得到了新型苯胺二聚体衍生物,从而解决了苯胺二聚体聚集诱导淬灭的问题,使其具有聚集诱导发光的特性。此外,本发明丰富了aie小分子的种类和制备方法,为新型aie体系的开发提供了一种新途径和新思路。
对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以对本发明的实施例做出若干变型和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!