一种螺环菌胺及其合成方法与流程
本发明涉及化合物合成领域,具体涉及一种螺环菌胺及其合成方法。
背景技术:
螺环菌胺,又名螺噁茂胺,英文名为spiroxamine,分子式是c18h35no2,分子量为297.476,cas登记号为118134-30-8,农用化学品一种。
螺环菌胺作用机理与特点:甾醇生物合成抑制剂,主要抑制c-14脱甲基化酶的合成。应用:适宜作物与安全性小麦和大麦。推荐剂量下对作物安全、无药害。防治对象小麦白粉病和各种锈病,大麦云纹病和条纹病。螺环菌胺是一种新型、内吸性的叶面杀菌剂,对白粉病特别有效;作用速度快且持效期长,兼具保护和治疗作用。既可以单独使用,又可以和其他杀菌剂混配以扩大杀菌谱。螺环菌胺在我国的市场前景广阔,因而其合成研究具有重要意义。
螺环菌胺以往的合成工艺路线是:首先对叔丁基苯酚钯炭催化加氢还原得到对叔丁基环已酮,再用对叔丁基环已酮与3-氯-1,2-丙二醇反应得到中间体8叔丁基-2-氯甲基-1,4-二氧杂螺(4,5)癸烷,最后再用中间体与n-乙基正丙胺胺化制得螺环菌胺,但是此种方法的成本较高,且螺环菌胺的反应收率较低,总收率为55%左右,不利于企业规模化生产。
技术实现要素:
针对现有技术中存在的问题,本发明的目的在于提供一种螺环菌胺及其合成方法,该螺环菌胺先合成含量在99%以上、转化率在95%以上的3-(n-乙基-n-正丙胺)-1,2-丙二醇,再利用3-(n-乙基-n-正丙胺)-1,2-丙二醇与对叔丁基环已酮合成螺环菌胺,合成方法简单,成本低廉,合成的螺环菌胺的含量达98.3%以上,收率达91%以上。
为了达到上述目的,本发明采用以下技术方案予以实现。
(一)一种螺环菌胺,包括以下制备原料:3-(n-乙基-n-正丙胺)-1,2-丙二醇、对叔丁基环已酮、二甲苯、浓硫酸、饱和氯化钠水溶液。
优选的,所述原料的用量为:3-(n-乙基-n-正丙胺)-1,2-丙二醇140-161份、对叔丁基环已酮135-161份、二甲苯550-650份、浓硫酸90-107份、饱和氯化钠水溶液150-250份。
进一步优选的,所述原料的用量为:3-(n-乙基-n-正丙胺)-1,2-丙二醇147份、对叔丁基环已酮141份、二甲苯600份、浓硫酸98份、饱和氯化钠水溶液200份。
(二)一种螺环菌胺的合成方法,包括以下步骤:
步骤1,合成3-(n-乙基-n-正丙胺)-1,2-丙二醇,备用;
步骤2,合成对叔丁基环已酮,备用;
步骤3,将所述3-(n-乙基正丙胺)-1,2-丙二醇、对叔丁基环已酮和二甲苯充分搅拌混合均匀,滴加浓硫酸,所述浓硫酸滴加完成后,升温回流脱水,降温,调节ph,静置分层,得上层有机相层和下层水层;
除去下层水层,向上层有机相层中加入饱和氯化钠水溶液洗一次,再采用水循环真空泵蒸馏去除溶剂、杂质和3-(n-乙基正丙胺)-1,2-丙二醇,最后采用旋片式真空泵进行减压蒸馏,得淡黄色油状液体螺环菌胺。
优选的,步骤1中,所述3-(n-乙基-n-正丙胺)-1,2-丙二醇的合成方法包含以下子步骤:
子步骤1.1,在氮气保护下,对环氧丙醇边搅拌边缓慢升温,升温至50~55℃时滴加n-乙基正丙胺,所述n-乙基正丙胺滴加结束后,反应,降温,得混合物;
子步骤1.2,向所述混合物中加入甲基异丁基酮和氯化钠饱和水溶液,搅拌,转入分液漏斗中静置分层,得下层水层和上层有机相层;
对所述下层水层再次进行萃取,得萃取有机相层;合并所述上层有机相层和所述萃取有机相层,得粗产品;
子步骤1.3,对所述粗产品进行减压蒸馏,得无色透明液体3-(n-乙基-n-正丙胺)-1,2-丙二醇。
进一步优选的,所述3-(n-乙基-n-正丙胺)-1,2-丙二醇的合成方法中,原料的用量为:环氧丙醇80-200份、n-乙基正丙胺120-250份、甲基异丁基酮450-1000份、氯化钠饱和水溶液100-400份。
进一步优选的,所述3-(n-乙基-n-正丙胺)-1,2-丙二醇的合成方法中,所述原料的用量为:环氧丙醇100份、n-乙基正丙胺124份、甲基异丁基酮500份、氯化钠饱和水溶液200份。
优选的,子步骤1.1中,所述n-乙基正丙胺的滴加时间为3.5-4.5小时。
优选的,子步骤1.1中,所述反应的温度为55-65℃,反应的时间为3.5-5.5小时。
优选的,子步骤1.1中,所述降温为降温至25-30℃。
优选的,子步骤1.2中,所述搅拌的时间为20-40分钟。
优选的,子步骤1.2中,对所述下层水层再次进行萃取采用甲基异丁基酮进行萃取。
优选的,子步骤1.3中,所述减压蒸馏的温度为90~101℃,减压蒸馏的真空度在500pa下,减压蒸馏的时间为2-5小时。
优选的,步骤2中,所述对叔丁基环已酮的合成方法包含以下子步骤:
子步骤2.1,将钯炭催化剂与甲苯混合,升温回流脱水,加入对叔丁基苯酚,氢化反应,降温至室温,过滤除去钯炭催化剂,得有机相;
子步骤2.2,采用水循环真空泵对所述有机相蒸馏去除甲苯,再用旋片式机械泵回收110~113℃下的副产物对叔丁基环已醇,最后回收115~117℃下的产物对叔丁基环已酮。
优选的,子步骤2.1中,所述钯炭催化剂、甲苯、对叔丁基苯酚的用量比为0.5-1.5:250-310:100-200。
优选的,子步骤2.1中,所述升温回流脱水的温度为110-118℃,升温回流脱水的时间为3-5小时。
优选的,子步骤2.1中,所述氢化反应的温度为150-154℃,氢化反应的压力为0.6-1.0mpa,氢化反应的时间为8-12小时。
优选的,步骤3中,所述浓硫酸的滴加时间为1.8-2.2小时。
优选的,步骤3中,所述升温回流脱水的温度为135-145℃,升温回流脱水的时间为12-16小时。
优选的,步骤3中,所述降温为降温至60-80℃。
优选的,步骤3中,所述调节ph为采用浓度为30%氢氧化钠溶液调节ph至7.5-8.5。
优选的,步骤3中,所述静置分层的时间为20-40分钟。
优选的,步骤3中,所述水循环真空泵的蒸馏温度为70~85℃,水循环真空泵的蒸馏时间为2.5-3小时,水循环真空泵的压力为-0.092mpa。
优选的,步骤3中,所述减压蒸馏的温度为105~108℃,减压蒸馏的时间为2-3小时,减压蒸馏的压力为300pa。
与现有技术相比,本发明的有益效果为:
(1)本发明的3-(n-乙基-n-正丙胺)-1,2-丙二醇是一种新的化合物,且通过gc-ms对实施例1中子步骤1.3收集99~101℃产物进行结构鉴定,确定了确定收集99~101℃产物为3-(n-乙基-n-正丙胺)-1,2-丙二醇;其合成方法简单,生产成本低,合成的3-(n-乙基-n-正丙胺)-1,2-丙二醇的含量在99%以上,转化率在95%以上。
(2)本发明利用利用3-(n-乙基-n-正丙胺)-1,2-丙二醇与对叔丁基环已酮合成螺环菌胺,合成方法简单,成本低廉,合成的螺环菌胺的含量达98.3%以上,收率达91%以上。
附图说明
下面结合附图和具体实施例对本发明做进一步详细说明。
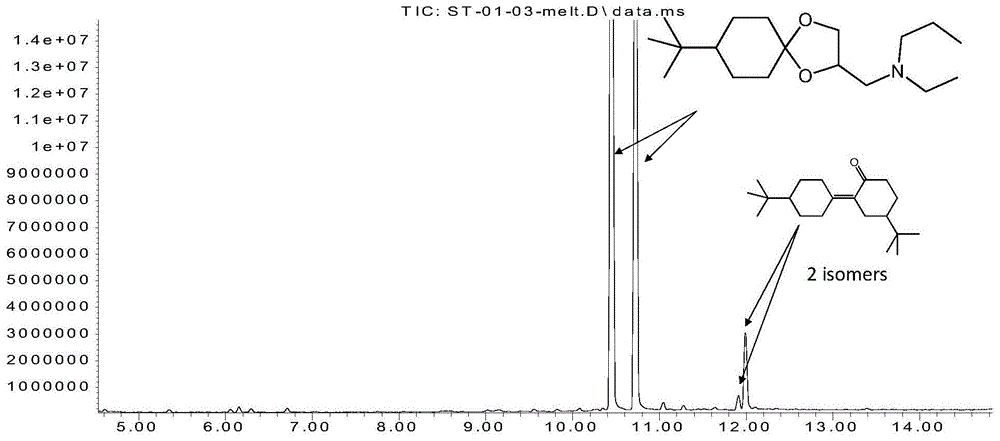
图1为本发明实施例1中步骤3所得的蒸馏后的产物的gc-ms图;
图2为图1中10.435min处的出峰点的质谱图;
图3为图1中10.709min处的出峰点的质谱图;
图4为图1中11.912min处的出峰点的质谱图;
图5为图1中11.986min处的出峰点的质谱图;
图6为本发明实施例1中步骤3所得的蒸馏后的残留物的gc-ms图;
图7为图6中5.346min处的出峰点的质谱图;
图8为图6中10.445min处的出峰点的质谱图;
图9为图6中10.732min处的出峰点的质谱图;
图10为图6中11.984min处的出峰点的质谱图;
图11为本发明实施例1中子步骤1.3收集99~101℃产物的gc-ms图;
图12为图11中6.192min处的出峰点的质谱图;
图13为图11中5.347min处的出峰点的质谱图;
图14为图11中7.023min处的出峰点的质谱图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域的技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。
实施例1
一种螺环菌胺的合成方法,包括以下步骤:
步骤1,合成3-(n-乙基-n-正丙胺)-1,2-丙二醇
具体的,步骤1包含以下子步骤:
子步骤1.1,向500ml的四口圆底烧瓶中,加入100g缩水干油(环氧丙醇),在氮气保护下,边搅拌边缓慢升温,控制温度在50~55℃之间滴加n-乙基正丙胺124g,约4小时滴加完。滴加结束后,升温至60℃反应4小时。反应结束后,降温至27℃,得混合物,转入1000ml三口圆底烧瓶中。
子步骤1.2,向混合物中加入甲基异丁基酮500g和氯化钠饱和水溶液200g,搅拌30分钟,转入1000ml分液漏斗中,静置分层,得下层水层和上层有机相层。分去下层水层,并采用50ml甲基异丁基酮对分去的水层再萃取一次,得到二次有机相;合并两次有机相,得到产物为粗产品。
子步骤1.3,采用旋片式真空泵对粗产品进行减压蒸馏,减压蒸馏的真空度在500pa下,气相在90℃下先蒸出未反应的n-乙基正丙胺、缩水甘油和溶剂甲基异丁基酮,然后减压蒸馏2小时收集99~101℃产物,得208g无色透明液体3-(n-乙基-n-正丙胺)-1,2-丙二醇,密度为0.958g/ml,含量为99.3%,转化率为95%。具体的3-(n-乙基-n-正丙胺)-1,2-丙二醇合成反应公式如下:
步骤2,合成对叔丁基环已酮
具体的,步骤2包含以下子步骤:
子步骤2.1,在500ml圆底烧瓶中加入钯炭催化剂0.71g,加入甲苯280ml,升温至114℃回流脱水4小时,将钯炭中水脱干净;然后转入到500ml的高压反应釜中,再加入150g的对叔丁基苯酚,在152℃、0.8mpa下氢化反应10小时;反应结束,降温到25℃,过滤除去钯炭催化剂,得到有机相。其中,有机相中包含甲苯、对叔丁基环已醇、对叔丁基环已酮和未完全反应的对叔丁基苯酚。
子步骤2.2,对有机相先用水循环真空泵在温度为45~55℃、压力为-0.092mpa条件下蒸馏1小时蒸除溶剂甲苯,然后用旋片式机械泵,用280mm填充柱蒸馏,20mmhg下,先在110~113℃下回收副产物对叔丁基环已醇(2小时基本回收完成),然后在115~117℃回收对叔丁基环已酮2小时,回收得到141g对叔丁基环已酮,含量为98.6%,收率为90%。具体对叔丁基环已酮的合成反应公式如下:
步骤3,合成螺环菌胺
在1000ml三口圆底烧瓶中加入3-(n-乙基正丙胺)-1,2-丙二醇147g、对叔丁基环已酮141g和二甲苯600ml,搅拌溶解后,常温下滴加浓硫酸98g,2小时加完后,缓慢升温至140℃回流脱水约14小时,脱出水15.3g后,降温至80℃。滴加30%氢氧化钠调ph值为8,搅拌30分钟不变后,转入分液漏斗中静置30分钟,得到上层有机相层和下层水层。
分去下层水相,有机相加入饱和氯化钠水溶液200g洗一次。有机相转入蒸馏瓶中,先用水循环真空泵在温度为70~85℃、压力为-0.092mpa条件下蒸馏2.5小时除掉溶剂二甲苯、两个未知杂质以及3-(n-乙基正丙胺)-1,2-丙二醇。然后用旋片式真空泵在压力为300pa、温度为105~108℃下减压蒸馏2小时,得蒸馏后的产物和残留物;其中,产物为253g淡黄色油状液体螺环菌胺,含量为98.3%,收率为91%。具体的螺环菌胺合成公式如下:
实施例2
一种螺环菌胺的合成方法,包括以下步骤:
步骤1,合成3-(n-乙基-n-正丙胺)-1,2-丙二醇
具体的,步骤1包含以下子步骤:
子步骤1.1,向500ml的四口圆底烧瓶中,加入120g缩水干油(环氧丙醇),在氮气保护下,边搅拌边缓慢升温,控制温度在50~55℃之间滴加n-乙基正丙胺150g,约4.5小时滴加完。滴加结束后,升温至65℃反应3.5小时。反应结束后,降温至25℃,得混合物,转入1000ml三口圆底烧瓶中。
子步骤1.2,向混合物中加入甲基异丁基酮625g和氯化钠饱和水溶液240g,搅拌35分钟,转入1000ml分液漏斗中,静置分层,得下层水层和上层有机相层。分去下层水层,并采用60ml甲基异丁基酮对分去的水层再萃取一次,得到二次有机相;合并两次有机相,得到产物为粗产品。
子步骤1.3,采用旋片式真空泵对粗产品进行减压蒸馏,减压蒸馏的真空度在500pa下,气相在90℃下先蒸出未反应的n-乙基正丙胺、缩水甘油和溶剂甲基异丁基酮,然后收集99~101℃产物,得251g无色透明液体3-(n-乙基-n-正丙胺)-1,2-丙二醇,含量为99.4%,转化率为96.20%。
步骤2,合成对叔丁基环已酮
具体的,步骤2包含以下子步骤:
子步骤2.1,在500ml圆底烧瓶中加入钯炭催化剂0.5g,加入甲苯250ml,升温至118℃回流脱水3.5小时,将钯炭中水脱干净;然后转入到500ml的高压反应釜中,再加入106g的对叔丁基苯酚,在154℃、0.8mpa下氢化反应8小时;反应结束,降温到25℃,过滤除去钯炭催化剂,得到有机相。其中,有机相中包含甲苯、对叔丁基环已醇、对叔丁基环已酮和未完全反应的对叔丁基苯酚。
子步骤2.2,对有机相先用水循环真空泵在温度为45~55℃、压力为-0.092mpa条件下蒸馏1小时蒸除溶剂甲苯,然后用旋片式机械泵,用280mm填充柱蒸馏,20mmhg下,先在110~113℃下回收副产物对叔丁基环已醇(2.2小时基本回收完成),然后在115~117℃回收对叔丁基环已酮1.8小时,回收得到对叔丁基环已酮99.0g,含量为98.9%,收率为90.5%。
步骤3,合成螺环菌胺
在1000ml三口圆底烧瓶中加入3-(n-乙基正丙胺)-1,2-丙二醇140g、对叔丁基环已酮135g和二甲苯600ml,搅拌溶解后,常温下滴加浓硫酸93g,1.8小时加完后,缓慢升温至145℃回流脱水约13.5小时,脱出水14.4g后,降温至75℃。滴加30%氢氧化钠调ph值为8,搅拌30分钟不变后,转入分液漏斗中静置30分钟,得到上层有机相层和下层水层。
分去下层水相,有机相加入饱和氯化钠水溶液200g洗一次。有机相转入蒸馏瓶中,先用水循环真空泵在温度为70~85℃、压力为-0.092mpa条件下蒸馏3小时蒸馏除掉溶剂二甲苯、两个未知杂质以及3-(n-乙基正丙胺)-1,2-丙二醇。然后用旋片式真空泵在压力为300pa、温度为105~108℃下减压蒸馏2.5小时,得蒸馏后的产物和残留物;其中,241g产物为淡黄色油状液体螺环菌胺,含量为98.6%,收率为91.7%。
实施例3
一种螺环菌胺的合成方法,包括以下步骤:
步骤1,合成3-(n-乙基-n-正丙胺)-1,2-丙二醇
具体的,步骤1包含以下子步骤:
子步骤1.1,向500ml的四口圆底烧瓶中,加入110g缩水干油(环氧丙醇),在氮气保护下,边搅拌边缓慢升温,控制温度在50~55℃之间滴加n-乙基正丙胺136g,约3.7小时滴加完。滴加结束后,升温至58℃反应4.2小时。反应结束后,降温至27℃,得混合物,转入1000ml三口圆底烧瓶中。
子步骤1.2,向混合物中加入甲基异丁基酮600g和氯化钠饱和水溶液200g,搅拌30分钟,转入1000ml分液漏斗中,静置分层,得下层水层和上层有机相层。分去下层水层,并采用75ml甲基异丁基酮对分去的水层再萃取一次,得到二次有机相;合并两次有机相,得到产物为粗产品。
子步骤1.3,采用旋片式真空泵对粗产品进行减压蒸馏,减压蒸馏的真空度在500pa下,气相在90℃下先蒸出未反应的n-乙基正丙胺、缩水甘油和溶剂甲基异丁基酮,然后收集99~101℃产物,得228g无色透明液体3-(n-乙基-n-正丙胺)-1,2-丙二醇,含量为99.37%,转化率为95.8%。
步骤2,合成对叔丁基环已酮
具体的,步骤2包含以下子步骤:
子步骤2.1,在500ml圆底烧瓶中加入钯炭催化剂0.8g,加入甲苯310ml,升温至110℃回流脱水3小时,将钯炭中水脱干净;然后转入到500ml的高压反应釜中,再加入170g的对叔丁基苯酚,在150℃、1.0mpa下氢化反应8小时;反应结束,降温到25℃,过滤除去钯炭催化剂,得到有机相。其中,有机相中包含甲苯、对叔丁基环已醇、对叔丁基环已酮和未完全反应的对叔丁基苯酚。
子步骤2.2,对有机相先用水循环真空泵在温度为45~55℃、压力为-0.092mpa条件下蒸馏1小时蒸除溶剂甲苯,然后用旋片式机械泵,用280mm填充柱蒸馏,20mmhg下,先在110~113℃下回收副产物对叔丁基环已醇(1.8小时基本回收完成),然后在115~117℃回收对叔丁基环已酮2.1小时,回收得到对叔丁基环已酮158g,含量为98.8%,收率为90.4%。
步骤3,合成螺环菌胺
在1000ml三口圆底烧瓶中加入3-(n-乙基正丙胺)-1,2-丙二醇161g、对叔丁基环已酮161g和二甲苯600ml,搅拌溶解后,常温下滴加浓硫酸107g,2.2小时加完后,缓慢升温135℃至回流脱水约16小时,脱出水16.6g后,降温至78℃。滴加30%氢氧化钠调ph值为8.2,搅拌30分钟不变后,转入分液漏斗中静置40分钟,得到上层有机相层和下层水层。
分去下层水相,有机相加入饱和氯化钠水溶液220g洗一次。有机相转入蒸馏瓶中,先用水循环真空泵在温度为70~85℃、压力为-0.092mpa条件下蒸馏3小时蒸馏除掉溶剂二甲苯、两个未知杂质以及3-(n-乙基正丙胺)-1,2-丙二醇。然后用旋片式真空泵在压力为300pa、温度为105~108℃下减压蒸馏3小时,得蒸馏后的产物和残留物;其中,273g产物为淡黄色油状液体螺环菌胺,含量为98.7%,收率为91.5%。
以上实施例中,子步骤1.2中,对下层水层可以进行多次萃取,以提高产物3-(n-乙基-n-正丙胺)-1,2-丙二醇的转换率。在螺环菌胺的合成过程中,先合成含量在99%以上、转化率在95%以上的3-(n-乙基-n-正丙胺)-1,2-丙二醇;再合成含量在98.6%、收率为90%以上的对叔丁基环已酮;最后利用合成的3-(n-乙基-n-正丙胺)-1,2-丙二醇与对叔丁基环已酮合成螺环菌胺,最终产物螺环菌胺的含量达98.3%以上、收率达91%以上;在合成螺环菌胺的过程中每一步的中间产物的收率均较高,可以显著降低螺环菌胺的生产成本。
采用气相色谱-质谱联用仪(gc-ms)对实施例1中步骤3所得的蒸馏后的产物和残留物分别进行结构鉴定,测试条件为:vf-5ms,30m,0,25mm,0,25μmfilm,100kpahe,2min100℃,20℃/min→260℃,i:250℃,蒸馏后的产物的测试结果如图1-5所示,蒸馏后的残留物的测试结果如图6-10所示,具体如下:
图1为本发明实施例1中步骤3所得的蒸馏后的产物的gc-ms图;对图1中10.435min、10.709min处的出峰点再次进行质谱分析,结果分别如图2、图3所示。对图2进行分析,结果如表1所示:
表1
由图2、图3和表1可知,在10.435min、10.709min处的出峰点的物质的结构如下:
由此可知,本发明实施例1中步骤3所得的蒸馏后的产物中主要含有螺环菌胺。
对图1中11.912min、11.986min处的出峰点再次进行质谱分析,结果分别如图4、图5所示。对图5进行分析,结果如表2所示:
表2
由图4、图5和表2可知,在11.912min、11.986min处的出峰点的物质的结构如下:
由此可知,本发明实施例1中步骤3所得的蒸馏后的产物中含有少量的5,4′-双环已烯-2-酮,此物质是对叔丁基环已酮与3-(n-乙基正丙胺)-1,2-丙二醇合成螺环菌胺时,两分子对叔丁基环已酮在硫酸下作用下生成的副产物。
图6为本发明实施例1所得的蒸馏后的残留物的gc-ms图;对图6中5.346min处的出峰点再次进行质谱分析,结果如图7所示。对图7进行分析,结果如表3所示:
表3
由表3可知,在5.346min处的出峰点的物质的结构如下:
由此可知,本发明实施例1中步骤3所得的蒸馏后的残留物中含有未完全反应的对叔丁基环已酮。
对图6中10.445min、10.732min处的出峰点再次进行质谱分析,结果分别如图8、图9所示。对图8进行分析,结果如表4所示:
表4
由表4、图8-9可知,在10.445min、10.732min处的出峰点的物质的结构如下:
由此可知,本发明实施例1中步骤3所得的蒸馏后的残留物中含有少量的螺环菌胺。
对图6中11.984min处的出峰点再次进行质谱分析,结果分别图10所示。对图10进行分析,结果如表5所示:
表5
由表5可知,在11.984min处的出峰点的物质的结构如下:
由此可知,本发明实施例1中步骤3所得的蒸馏后的残留物中含有残留中含有6%的5,4′-双环已烯-2-酮。
综上所述,本发明通过对步骤3中蒸馏后的产物采用gc-ms进行结构鉴定,确定其产物就是螺环菌胺,并通过对蒸馏后的残留物进行gc-ms分析,再次验证本发明最终合成的物质为螺环菌胺。
采用气相色谱-质谱联用仪(gc-ms)对实施例1中子步骤1.3收集99~101℃产物进行结构鉴定,测试条件为:vf-5ms,30m,0,25mm,0,25μmfilm,100kpahe,2min100℃,20℃/min→260℃,i:250℃,测试结果如图11-14所示,具体如下:
图11为本发明实施例1中子步骤1.3收集99~101℃产物的gc-ms图;对图11中6.192min处的出峰点再次进行质谱分析,结果如图12所示。对图12进行分析,结果如表6所示:
表6
由表6可知,在6.192min处的出峰点的物质的结构如下:
由此可知,本发明实施例1中子步骤1.3收集99~101℃产物中含有3-(n-乙基-n-正丙胺)-1,2-丙二醇。
对图11中5.347min、7.023min处的出峰点分别再次进行质谱分析,结果分别如图13、图14所示;并不清楚在5.347min、7.023min处的物质具体是什么。
因此,由图11-14可知,本发明中实施例1中子步骤1.3收集99~101℃产物中主要物质为3-(n-乙基-n-正丙胺)-1,2-丙二醇。
综上所述,本发明通过对子步骤1.3收集99~101℃产物采用gc-ms进行结构鉴定,确定其产物就是3-(n-乙基-n-正丙胺)-1,2-丙二醇;并通过对蒸馏后的残留物进行gc-ms分析(图未给出),再次验证本发明子步骤1.3合成的物质为3-(n-乙基-n-正丙胺)-1,2-丙二醇。
虽然,本说明书中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!