一种荜茇酰胺衍生物及其制备方法和用途与流程
[0001]
本发明涉及医药化学技术领域,特别是涉及一种荜茇酰胺衍生物及其制备方法和用途。
背景技术:
[0002]
荜茇酰胺(piperlongumine,图1)是从胡椒科植物根茎中提取到的一种生物碱,具有抗肿瘤、抗抑郁、镇痛等多种药理作用。2011年哈佛大学schreiber教授研究组报道了荜茇酰胺优异的抗癌活性和通过诱导活性氧(ros)产生从而选择性杀死癌细胞的机制(nature 2011,475,231),该成果发表在《nature》上,引起了极大的反响,使荜茇酰胺近年来大有发展为抗癌明星分子的趋势。2013年诺贝尔生理医学奖得主jim watson教授又提出了一个关于ros非常重要的观点:“用于直接杀死癌细胞的绝大多数化疗试剂、放射疗法及部分靶向性药物,都是通过直接或间接地促进ros的生成来阻滞癌细胞生长周期中的关键步骤”(open biol.2013,3,120144)。这一观点的提出迅速开启了基于促氧化策略设计抗癌试剂的新热潮。细胞内氧化还原的平衡同时依赖于ros的产生及胞内抗氧化防御体系对ros的清除,而促氧化策略是基于这样一个事实:癌细胞为了维持其恶性增殖、侵袭、血管生成和转移等恶性表型,相比于正常细胞有着更高的固有ros水平及异常的氧化还原状态(antioxid.redox signal.2013,19:854-895)。因此,癌细胞相比于正常细胞,对氧化应激水平的进一步升高或抗氧化体系的有效抑制显得更加敏感,也更容易达到其死亡阈值。基于以上差异,癌细胞的氧化还原缺陷可能作为癌症预防和治疗的一个重要靶点。目前获美国食品和药品监督管理局的批准认可的抗癌药物中,已有许多药物(伊利司莫、2-甲氧雌二醇与顺铂等)涉及到促进ros生成的作用机制。
[0003]
上述事实足以证明基于促氧化的抗癌策略的重要性,基于上述出发点,抗氧化防御体系中重要蛋白硫氧还蛋白还原酶(trxr)成为我们关注的焦点。原因如下:(1)硫氧还蛋白系统是细胞内最主要的氧化还原系统之一,对维持胞内氧化还原平衡起着关键的作用,主要由烟酰胺腺嘌呤二核苷酸磷酸(nadph)、trxr和硫氧还蛋白(trx)三者组成。trx被nadph还原后将电子进一步传递给底物,进而参与胞内更多的氧化还原事件,而trxr是目前已知的唯一可以催化trx还原的酶(free radical bio.med.2014,66:75-87)。(2)肿瘤组织中trxr的水平远远高于正常组织。因此我们相信,trxr是基于ros与耐药性的癌治疗策略的有效靶点。
[0004]
事实上,大多靶向氧化还原敏感蛋白的化合物都具有一个结构共性,即含有michael加成受体(-不饱和酮)结构单元,它们作为亲电体可以同胞内氧化还原敏感的巯基蛋白或硒蛋白共价加成。哺乳动物体内三种trxr异构酶的碳末端(-gly-cys-secys-gly-)都含有一个硒代半胱氨酸(secys)残基,这是trxr发挥抗氧化酶活性的重要元件。如图1中姜黄素(curcumin)、紫草素(shikonin)、黄腐酚(xanthohumol)、利尿酸(ethacrynic acid)及环烯酮类(cycloalkenones)等亲电试剂可与trxr活性位点的硒半胱氨酸负离子发生共价反应,并且这种不可逆的共价修饰不仅会抑制trxr的活性,还会使trxr修饰物表现出诱
导nadph氧化的活性进而促进ros的产生,成为ros的重要来源(j.med.chem.2016,59:1747
–
1760;j.med.chem.2015,58:1795-1805)。荜茇酰胺结构中也包含两个michael加成受体单元(图1),但目前并没有研究证实trxr也可以作为一个重要靶点贡献荜茇酰胺优越的促氧化活性。综上所述,从促氧化角度出发设计荜茇酰胺导向的新型抗癌先导化合物,并探究它们的作用机制与结构基础,成为一项迫切而具有吸引力的工作。这一工作的顺利开展也将为天然产物介导的新药开发提供有价值的基础资料。
技术实现要素:
[0005]
本发明的目的在于克服上述现有技术的不足,提供一种荜茇酰胺衍生物及其制备方法和用途,该荜茇酰胺类衍生物含有多个michael加成受体单元,其新颖的结构,提高了其亲电性,使其具有很好的蛋白靶向潜力,其对硫氧还蛋白还原酶有很好的抑制作用;在抗肿瘤生物活性体外筛选实验中,该类衍生物对肿瘤细胞的生长也出现了一定的抑制作用,同时该类衍生物在a549人肺癌细胞中表现出促进活性氧生成的作用。
[0006]
为了实现上述目的,本发明采用如下技术方案:
[0007]
一种荜茇酰胺衍生物,所述荜茇酰胺衍生物具有以下结构:
[0008]
其中r1、r2、r3、r4选自h、卤素、三氟甲基、c
1-c3烷基、c
1-c3烷氧基中的任意一种。卤素是指氟、氯、溴、碘;c
1-c3烷基是指甲基、乙基、丙基、异丙基;c
1-c3烷氧基是指甲氧基、乙氧基、丙氧基、异丙氧基。
[0009]
其中r1、r2、r3、r4选自h、f、cl、三氟甲基、甲基、甲氧基中的任意一种。
[0010]
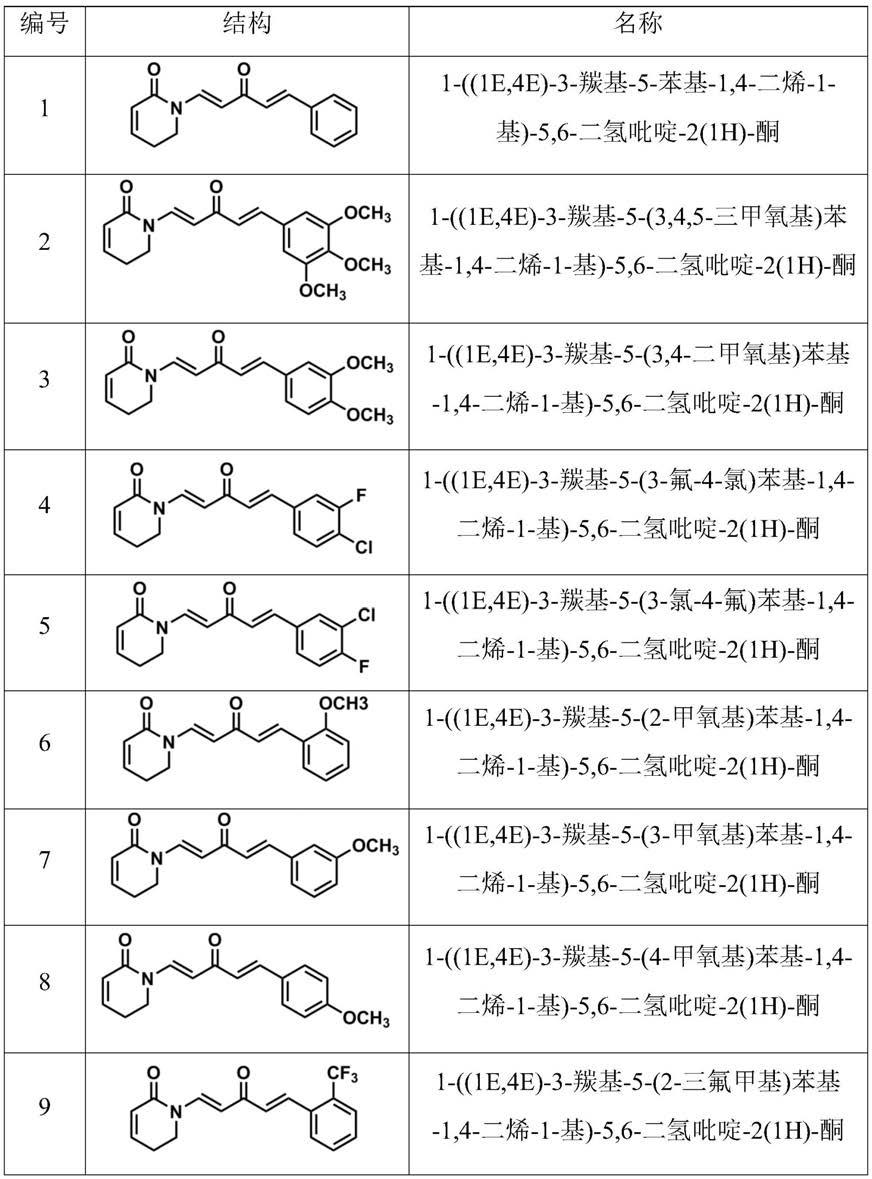
所述荜茇酰胺衍生物包括但不限于1-((1e,4e)-3-羰基-5-苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3,4,5-三甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3,4-二甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3-氟-4-氯)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3-氯-4-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(2-甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3-甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(4-甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(2-三氟甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3-三氟甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(4-三氟甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(2-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(4-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(2-甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(3-甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮、1-((1e,4e)-3-羰基-5-(4-甲
基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮。上述荜茇酰胺衍生物的结构式如表1所示。
[0011]
表1:
[0012]
[0013][0014]
一种荜茇酰胺衍生物的制备方法,包括以下步骤:以3-丁烯胺盐酸盐为起始原料,和三乙胺、2-丁烯酰氯反应生成(e)-n-(丁-3-烯-1-基)丁-2-烯酰胺;所得(e)-n-(丁-3-烯-1-基)丁-2-烯酰胺在grubbs第二代催化剂作用下反应生成5,6-二氢吡啶-2(1h)-酮;5,6-二氢吡啶-2(1h)-酮与甲酸乙酯反应得到n-甲酰基-5,6-二氢吡啶-2(1h)-酮;苯甲醛或其取代物与丙酮或1-三苯基膦-2-丙酮反应生成苯基上各种取代的(e)-4-苯基丁-3-烯-2-酮;5,6-二氢吡啶-2(1h)-酮与苯基上各种取代的(e)-4-苯基丁-3-烯-2-酮反应制得荜茇酰胺衍生物。
[0015]
荜茇酰胺衍生物的制备方法,具体包括以下步骤:
[0016]
(1)将3-丁烯胺盐酸盐1.0equiv溶于5.0ml无水二氯甲烷中,在0℃、n2保护下搅拌,并依次加入三乙胺2.0equiv、dmap 0.2equiv、2-丁烯酰氯1.2equiv,室温反应2小时,然后加入50ml二氯甲烷和20ml饱和碳酸氢钠水溶液,有机层用无水硫酸钠干燥,经过滤、减压浓缩、柱层析分离得到(e)-n-(丁-3-烯-1-基)丁-2-烯酰胺;
[0017]
(2)将步骤(1)制得的(e)-n-(丁-3-烯-1-基)丁-2-烯酰胺1.0equiv溶于120ml无
水二氯甲烷中,加入grubbs第二代催化剂0.025equiv,搅拌加热回流2小时,减压旋除溶剂,柱层析分离,得到5,6-二氢吡啶-2(1h)-酮;
[0018]
(3)在高温干燥过的圆底烧瓶中加入步骤(2)制得的5,6-二氢吡啶-2(1h)-酮1.0equiv、甲酸乙酯3.0equiv、三乙胺0.24equiv和20ml二氯甲烷,在0℃时加入四氯化钛2.0equiv,使反应体系在0℃下继续反应1小时,再用50ml水淬灭,30ml乙酸乙酯萃取三次,结合有机相用无水硫酸钠干燥,柱层析分离,得到n-甲酰基-5,6-二氢吡啶-2(1h)-酮;
[0019]
(4)在烧瓶中依次加入苯甲醛或其取代物1.0equiv、丙酮或1-三苯基膦-2-丙酮1.6equiv,用二氯甲烷溶解,再将反应体系从室温转移至90℃油浴中,继续反应1小时,经柱层析分离,得到苯基上各种取代的(e)-4-苯基丁-3-烯-2-酮;
[0020]
(5)在一个装有磁搅拌棒的火焰干燥的25ml圆底烧瓶中加入三聚氰氯1.0equiv,在室温下滴加6.0equiv的n-甲酰基-5,6-二氢吡啶-2(1h)-酮5分钟,转移到室温油浴加热,在100℃搅拌反应两个小时,反应后移除油浴并冷却到室温,得到红橙色的固体;向含有红橙色固体的反应容器中加入2ml四氢呋喃,将混合物加热回流,直到固体完全溶解,加入6ml己烷,将混合物缓慢冷却到室温,然后用己烷/四氢呋喃混合溶剂进行洗涤,得到红色固体;将步骤(4)制得的(e)-4-苯基丁-3-烯-2-酮1.0equiv和红色固体1.1equiv在65℃下加热反应12小时,经柱层析分离,得到荜茇酰胺衍生物。
[0021]
步骤(2)中,搅拌加热回流温度为40℃;步骤(5)中,混合物加热回流温度为70℃。
[0022]
步骤(4)中,苯甲醛取代物为苯甲醛、3,4,5-三甲氧基苯甲醛、3,4-二甲氧基苯甲醛、3-氟-4-氯苯甲醛、3-氯-4-氟苯甲醛、2-甲氧基苯甲醛、3-甲氧基苯甲醛、4-甲氧基苯甲醛、2-三氟甲基苯甲醛、3-三氟甲基苯甲醛、4-三氟甲基苯甲醛、2-氟苯甲醛、3-氟苯甲醛、4-氟苯甲醛、2-甲基苯甲醛、3-甲基苯甲醛或4-甲基苯甲醛。
[0023]
步骤(5)中,己烷/四氢呋喃混合溶剂中己烷与/四氢呋喃体积比为2:1。
[0024]
步骤(1)中,柱层析时以pe和ea体积比为1:1的混合液为洗脱溶剂;步骤(2)中,柱层析时以ea为洗脱溶剂;步骤(3)中,柱层析时以pe和ea体积比为3:1-5:1的混合液为洗脱溶剂;步骤(4)中,柱层析时以pe和ea体积比为5:1的混合液为洗脱溶剂;步骤(5)中,柱层析时以pe和ea体积比为10:7-5:2的混合液为洗脱溶剂。
[0025]
本发明以天然产物荜茇酰胺为先导化合物对其进行结构改造,通过上述制备方法在荜茇酰胺结构中增加一个双键,得到含戊二烯酮结构(对称的双michael加成位点)的骨架,提供一种含有三个michael加成受体单元荜茇酰胺类衍生物,制备荜茇酰胺衍生物的反应路线如图2所示。
[0026]
所述荜茇酰胺衍生物可用于制备靶向硫氧还蛋白还原酶的促氧化抗癌药物。
[0027]
本发明的有益效果是:(1)本发明提供的新型荜茇酰胺类衍生物对硫氧还蛋白还原酶有很好的抑制作用;(2)在抗肿瘤生物活性体外筛选实验中,该类衍生物对肿瘤细胞的生长也出现了一定的抑制作用,同时该类衍生物在a549人肺癌细胞中表现出促进活性氧生成的作用;(3)本发明提供的含有多个michael加成受体单元的荜茇酰胺类衍生物,其新颖的结构,提高了其亲电性,使其具有很好的蛋白靶向潜力。
附图说明
[0028]
图1为本发明的含michael加成受体结构单元的活性分子的结构式;
[0029]
图2为本发明的制备荜茇酰胺衍生物的反应路线图;
[0030]
图3为本发明的体现实施例2制备的荜茇酰胺衍生物诱导a549细胞内活性氧水平的测试结果对比图;
[0031]
图4为本发明的体现实施例1-3制备的荜茇酰胺衍生物抑制trxr的活性的测试结果对比图。
具体实施方式
[0032]
下面结合附图和具体实施方式对本发明作进一步描述:
[0033]
实施例1
[0034]
一种荜茇酰胺衍生物,具有以下结构:
[0035][0036]
该荜茇酰胺衍生物的制备方法,包括以下步骤:
[0037]
(1)3-丁烯胺盐酸盐1.0equiv溶于5.0ml无水二氯甲烷中,在0℃,n2保护下搅拌,并依次加入三乙胺2.0equiv、dmap 0.2equiv、2-丁烯酰氯1.2equiv,室温反应2小时,加入50ml二氯甲烷和20ml饱和碳酸氢钠水溶液,有机层用无水硫酸钠干燥,经过滤、减压浓缩、柱层析(pe/ea=1:1)分离,得到(e)-n-(丁-3-烯-1-基)丁-2-烯酰胺,其产率为85%;
[0038]
(2)将步骤(1)制得的(e)-n-(丁-3-烯-1-基)丁-2-烯酰胺1.0equiv溶于120ml无水二氯甲烷中,加入grubbs第二代催化剂0.025equiv,于40℃搅拌加热回流2小时,减压旋除溶剂,经柱层析(ea)分离,得到5,6-二氢吡啶-2(1h)-酮,其产率为56%;
[0039]
(3)在高温干燥过的圆底烧瓶中加入步骤(2)制得的5,6-二氢吡啶-2(1h)-酮1.0equiv、甲酸乙酯3.0equiv、三乙胺0.24equiv和20ml二氯甲烷,在0℃时加入四氯化钛2.0equiv,使反应体系在0℃下继续反应1小时,再用50ml水淬灭,30ml乙酸乙酯萃取三次,结合有机相用无水硫酸钠干燥,柱层析(pe/ea=3:1-5:1)分离,得到n-甲酰基-5,6-二氢吡啶-2(1h)-酮,其产率为76%;
[0040]
(4)在烧瓶中依次加入苯甲醛1.0equiv、丙酮1.6equiv,用少量二氯甲烷溶解,再将反应体系从室温转移至90℃油浴中,继续反应1小时,柱层析(pe/ea=5:1),得到(e)-4-苯基丁-3-烯-2-酮,其产率为82%;
[0041]
(5)在一个装有磁搅拌棒的火焰干燥的25毫升圆底烧瓶中加入三聚氰氯1.0equiv,在室温下滴加6.0equiv的n-甲酰基-5,6-二氢吡啶-2(1h)-酮5分钟,转移到室温油浴加热,在100℃搅拌反应两个小时,移除油浴和冷却到室温,得到红橙色的固体;向含有红橙色固体的反应容器中加入2ml四氢呋喃,于70℃将混合物加热回流,直到固体完全溶解,加入6ml己烷,将混合物缓慢冷却到室温,然后用己烷/四氢呋喃混合溶剂(己烷与四氢呋喃体积比为2:1)进行洗涤,得到红色固体;将步骤(4)制得的(e)-4-苯基丁-3-烯-2-酮1.0equiv和红色固体1.1equiv在65℃下加热12小时,经柱层析(pe/ea=10:7-5:2)分离,得产物1-((1e,4e)-3-羰基-5-苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,记为化合物1,其产率为42%。
[0042]
荜茇酰胺衍生物结构通过核磁共振(nmr)或质谱(ms)来确定。nmr位移以百万分之一(ppm)的单位给出。nmr的测定是用bruker avance-400mhz核磁共振仪,测定溶剂为氘代氯仿(cdcl3),内标为四甲基硅烷(tms)。
[0043]1h nmr(400mhz,cdcl3):δ2.461(m,2h),3.452(m,2h),5.943(d,j=8.0hz,1h),6.011(d,j=8.0hz,1h),6.661(d,j=16.0hz,1h),7.104(d,j=16.0hz,1h),7.194(d,j=16.0hz,1h),7.305(m,1h),7.414(m,2h),7.540(m,2h),8.333(d,j=16.0hz,1h)。
[0044]
实施例2
[0045]
一种荜茇酰胺衍生物,具有以下结构:
[0046][0047]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了3,4,5-三甲氧基苯甲醛,将丙酮换成1-三苯基膦-2-丙酮,其反应条件及物料摩尔量均不变,得到(e)-4-(3,4,5-三甲氧基苯基)丁-3-烯-2-酮,产率为75%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(3,4,5-三甲氧基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(3,4,5-三甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,记为化合物2,产率为35%。
[0048]1h nmr(400mhz,cdcl3):δ2.451(m,2h),3.404(m,2h),3.750(m,1h),3.810(s,6h),6.034(d,j=8.0hz,1h),6.122(d,j=8.0hz,1h),6.608(d,j=16.0hz,1h),6.727(s,2h),7.138(d,j=16.0hz,1h),7.225(d,j=16.0hz,1h),8.358(d,j=16.0hz,1h)。
[0049]
实施例3
[0050]
一种荜茇酰胺衍生物,具有以下结构:
[0051][0052]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了3,4-二甲氧基苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(3,4-二甲氧基苯基)丁-3-烯-2-酮,产率为65%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(3,4-二甲氧基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(3,4-二甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,记为化合物3,产率为37%。
[0053]1h nmr(400mhz,cdcl3):δ2.480(m,2h),3.365(m,2h),3.854(s,6h),5.955(d,j=8.0hz,1h),6.082(d,j=8.0hz,1h),6.567(d,j=16.0hz,1h),6.880(d,1h),7.087(s,1h),7.118(d,j=16.0hz,1h),7.149(m,1h),7.245(d,j=16.0hz,1h),8.384(d,j=16.0hz,1h)。
[0054]
实施例4
[0055]
一种荜茇酰胺衍生物,具有以下结构:
[0056][0057]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了3-氟-4-氯苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(3-氟-4-氯苯基)丁-3-烯-2-酮,产率为60%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(3-氟-4-氯苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(3-氟-4-氯)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为32%。
[0058]1h nmr(400mhz,cdcl3):δ2.302(m,2h),3.304(m,2h),6.054(d,j=8.0hz,1h),6.185(d,j=8.0hz,1h),6.211(d,j=16.0hz,1h),6.921(d,1h),7.254(d,j=16.0hz,1h),7.320(m,1h),7.458(m,1h),7.564(d,j=16.0hz,1h),8.352(d,j=16.0hz,1h)。
[0059]
实施例5
[0060]
一种荜茇酰胺衍生物,具有以下结构:
[0061][0062]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了3-氯-4-氟苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(3-氯-4-氟苯基)丁-3-烯-2-酮,产率为78%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(3-氯-4-氟苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(3-氯-4-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为40%。
[0063]1h nmr(400mhz,cdcl3):δ2.324(m,2h),3.351(m,2h),5.896(d,j=8.0hz,1h),6.225(d,j=16.0hz,1h),6.301(d,j=8.0hz,1h),6.981(d,1h),7.054(d,j=16.0hz,1h),7.322(m,1h),7.478(m,1h),7.504(d,j=16.0hz,1h),8.282(d,j=16.0hz,1h)。
[0064]
实施例6
[0065]
一种荜茇酰胺衍生物,具有以下结构:
[0066][0067]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了2-甲氧基苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(2-甲氧基苯基)丁-3-烯-2-酮,产率为60%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(2-甲氧基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(2-甲氧基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为43%。
[0068]1h nmr(400mhz,cdcl3):δ2.284(m,2h),3.301(m,2h),3.901(s,3h),6.016(d,j=8.0hz,1h),6.225(d,j=16.0hz,1h),6.877(d,j=8.0hz,1h),6.905(d,j=16.0hz,1h),7.012(m,1h),7.081(m,1h),7.402(m,1h),7.658(m,1h),8.014(d,j=16.0hz,1h),8.372
1-基)-5,6-二氢吡啶-2(1h)-酮,产率为32%。
[0083]1h nmr(400mhz,cdcl3):δ2.308(m,2h),3.365(m,2h),6.186(d,j=8.0hz,1h),6.305(d,j=16.0hz,1h),7.084(d,j=8.0hz,1h),7.125(d,j=16.0hz,1h),7.118(m,1h),7.277(m,1h),7.384(m,1h),7.468(m,1h),8.025(d,j=16.0hz,1h),8.402(d,j=16.0hz,1h)。
[0084]
实施例10
[0085]
一种荜茇酰胺衍生物,具有以下结构:
[0086][0087]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了3-三氟甲基苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(3-三氟甲基苯基)丁-3-烯-2-酮,产率为58%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(3-三氟甲基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(3-三氟甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为30%。
[0088]1h nmr(400mhz,cdcl3):δ2.281(m,2h),3.338(m,2h),6.143(d,j=8.0hz,1h),6.265(d,j=16.0hz,1h),7.014(d,j=8.0hz,1h),7.117(d,j=16.0hz,1h),7.139(m,1h),7.467(m,1h),7.489(m,1h),7.648(m,1h),7.896(d,j=16.0hz,1h),8.376(d,j=16.0hz,1h)。
[0089]
实施例11
[0090]
一种荜茇酰胺衍生物,具有以下结构:
[0091][0092]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了4-三氟甲基苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(4-三氟甲基苯基)丁-3-烯-2-酮,产率为58%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(4-三氟甲基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(4-三氟甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为30%。
[0093]1h nmr(400mhz,cdcl3):δ2.311(m,2h),3.408(m,2h),6.187(d,j=8.0hz,1h),6.298(d,j=16.0hz,1h),7.134(d,j=8.0hz,1h),7.198(d,j=16.0hz,1h),7.499(d,2h),7.586(d,2h),7.905(d,j=16.0hz,1h),8.390(d,j=16.0hz,1h)。
[0094]
实施例12
[0095]
一种荜茇酰胺衍生物,具有以下结构:
[0096]
[0097]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了2-氟苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(2-氟苯基)丁-3-烯-2-酮,产率为50%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(2-氟苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(2-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为28%。
[0098]1h nmr(400mhz,cdcl3):δ2.218(m,2h),3.255(m,2h),6.066(d,1h),6.205(d,j=16.0hz,1h),6.875(d,j=16.0hz,1h),6.984(d,1h),7.146(m,2h),7.157(m,1h),7.342(m,1h),8.115(d,j=16.0hz,1h),8.382(d,j=16.0hz,1h)。
[0099]
实施例13
[0100]
一种荜茇酰胺衍生物,具有以下结构:
[0101][0102]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了3-氟苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(3-氟苯基)丁-3-烯-2-酮,产率为50%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(3-氟苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(3-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为28%。
[0103]1h nmr(400mhz,cdcl3):δ2.248(m,2h),3.255(m,2h),5.986(d,1h),6.085(d,j=16.0hz,1h),6.985(m,1h),6.991(m,1h),7.014(d,1h),7.075(d,j=16.0hz,1h),7.286(m,1h),7.397(m,1h),7.924(d,j=16.0hz,1h),8.327(d,j=16.0hz,1h)。
[0104]
实施例14
[0105]
一种荜茇酰胺衍生物,具有以下结构:
[0106][0107]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了4-氟苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(4-氟苯基)丁-3-烯-2-酮,产率为55%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(4-氟苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(4-氟)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为31%。
[0108]1h nmr(400mhz,cdcl3):δ2.241(m,2h),3.338(m,2h),6.027(d,j=8.0hz,1h),6.128(d,j=16.0hz,1h),7.024(d,j=8.0hz,1h),7.058(d,j=16.0hz,1h),7.400(d,2h),7.687(d,2h),7.835(d,j=16.0hz,1h),8.334(d,j=16.0hz,1h)。
[0109]
实施例15
[0110]
一种荜茇酰胺衍生物,具有以下结构:
[0111][0112]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了2-甲基苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(2-甲基苯基)丁-3-烯-2-酮,产率为68%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(2-甲基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(2-甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为34%。
[0113]1h nmr(400mhz,cdcl3):δ2.310(m,2h),2.498(s,3h),3.445(m,2h),6.127(d,1h),6.295(d,j=16.0hz,1h),6.813(d,j=16.0hz,1h),6.989(m,1h),7.098(d,1h),7.137(m,1h),7.286(m,1h),7.312(m,1h),8.096(d,j=16.0hz,1h),8.355(d,j=16.0hz,1h)。
[0114]
实施例16
[0115]
一种荜茇酰胺衍生物,具有以下结构:
[0116][0117]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了3-甲基苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(3-甲基苯基)丁-3-烯-2-酮,产率为70%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(3-甲基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(3-甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为40%。
[0118]1h nmr(400mhz,cdcl3):δ2.357(m,2h),2.427(s,3h),3.402(m,2h),5.986(d,1h),6.135(d,j=16.0hz,1h),6.896(m,1h),7.014(d,1h),7.038(d,1h),7.168(s,1h),7.196(m,1h),7.583(m,1h),7.964(d,j=16.0hz,1h),8.343(d,j=16.0hz,1h)。
[0119]
实施例17
[0120]
一种荜茇酰胺衍生物,具有以下结构:
[0121][0122]
制备方法与实施例1相同,只是将第(4)歩中苯甲醛换成了4-甲基苯甲醛,其反应条件及物料摩尔量均不变,得到(e)-4-(4-甲基苯基)丁-3-烯-2-酮,产率为72%。将第(5)歩中(e)-4-苯基丁-3-烯-2-酮换成了(e)-4-(4-甲基苯基)丁-3-烯-2-酮,其反应条件及物料摩尔量均不变,得到1-((1e,4e)-3-羰基-5-(4-甲基)苯基-1,4-二烯-1-基)-5,6-二氢吡啶-2(1h)-酮,产率为35%。
[0123]1h nmr(400mhz,cdcl3):δ2.301(m,2h),2.462(s,3h),3.412(m,2h),6.098(d,1h),6.265(d,j=16.0hz,1h),7.092(d,1h),7.208(d,j=16.0hz,1h),7.386(d,2h),7.568(d,2h),7.912(d,j=16.0hz,1h),8.378(d,j=16.0hz,1h)。
[0124]
实施例18
[0125]
抗癌活性:以抗癌试剂姜黄素作为阳性对照,采用mtt法通过酶标仪分别测定实施例1-17制备的荜茇酰胺衍生物、姜黄素对癌细胞(人肝癌细胞hepg2、smmc-7721,人肺癌细胞a549、nci-h460)的增殖抑制活性。癌细胞均购于中科院上海细胞库。细胞在rpmi-1640培养基中,置于5%co2含量及37℃恒定温度的细胞培养箱中培养,每升培养基加入碳酸氢钠2g、链霉素青霉素各100ku,无菌过滤后添加10%胎牛血清。细胞以3
×
103个/孔的密度种于96孔板中,24h后加药处理48h,药物浓度梯度为0、5、10、15、20、25、30、40、50m,同时设定不加药处理的对照孔。更换新鲜培养基,加入mtt避光孵育4h,弃去mtt并加入dmso(100μl/孔)震荡后通过微孔板检测系统(bio-rad model 550酶标仪)在570nm处测定吸光度值。细胞存活率表示为相对于对照组的百分比。以药物浓度及对细胞存活率作图可得到剂量反应曲线,从中求出药物的半数抑制浓度(ic
50
)。实验重复3次。实验结果参见表1。
[0126]
表1本发明化合物的癌细胞增殖抑制活性
[0127][0128]
结果表明:多数荜茇酰胺衍生物显示出较好的细胞增殖抑制活性,部分荜茇酰胺衍生物表现出对a549细胞较好的增殖抑制选择性。
[0129]
实施例19
[0130]
促进ros生成的能力:采用荧光探针2',7'-二氯荧光黄通过流式细胞仪测定癌细胞增殖抑制活性较好的荜茇酰胺类衍生物处理后癌细胞内ros的水平。荧光探针dcfh-da具有细胞膜渗透性,它进入细胞后可在细胞酯酶的作用下水解得到不可透膜的dcfh,水解产物被胞内ros氧化生成dcf。dcf是荧光物质,其荧光强度的大小就可反映细胞内ros的浓度。将a549细胞以3
×
105个/孔的密度种至六孔板中,于37℃细胞培养箱培养24小时后换液,加入化合物2(化合物2的浓度分别为1、2、5μm),药物作用4小时或8小时后吸出培养基,加入胰蛋白酶消化并分别收集各孔细胞于2ml离心管中,250g离心6分钟除去培养基后用pbs(1ml/
次)以相同转速离心洗1-2次,然后加入dcfh-da染料(终浓度为3μm),置于37℃温箱中避光染色半小时,用pbs洗去残余染料后通过流式细胞仪进行检测。所测荧光的激发波为488nm,发射波长为525nm,同时设定不含化合物2的对照组。采用上述相同的方法测定姜黄素对细胞内ros生成的影响(姜黄素浓度5μm)。也采用相同的方法测定抗氧化剂对化合物2诱导细胞内ros生成的影响,抗氧化剂处理组需要提前加入nac(10mm)预处理1小时,再加入5μm的化合物2孵育8小时,同时设定抗氧化剂处理后不加药的对照组。上述结果相对于对照组进行归一化处理,每组实验均独立重复3次,实验结果参见图2。
[0131]
结果表明:化合物2能显著地诱导a549细胞中ros的产生,并且呈现出明显的浓度和时间依赖关系。在5μm的浓度下作用8h可将ros的水平提升到对照组水平的将近4倍,相比之下,姜黄素(5μm)在作用8h后仍未导致ros水平的升高。并且抗氧化剂nac可明显地逆转5μm的化合物2所诱导的ros产生,使dcf荧光强度几乎完全降回到了对照组的水平。
[0132]
实施例20
[0133]
trxr靶向能力:细胞外trxr的活性可采用dtnb法进行测定。将nadph(200m)还原处理5min的trxr(120nm)分别与化合物1、化合物2、化合物3(化合物1、化合物2、化合物3的浓度均为10m)混合于tris-edta缓冲液,并于室温下孵育1h,同时设定不含化合物2的对照组(记为空白对照组)。随后将100l上述混合液加入到800l工作液(2.25mm的dtnb与200m的nadph溶于tris-edta缓冲液)中,立即监测其在412nm处的吸光度值变化(升高),记录90秒,以δod对时间t作图求出斜率。通过各组所得斜率与空白对照组斜率相比,即可计算得到trxr抑制活性,表示为相对于空白对照组所占的百分比。以同样方法测定trxr抑制剂姜黄素的活性作为对照。上述每组实验均独立重复3次,实验结果参见图4。
[0134]
结果表明:10m的本发明化合物1、化合物2、化合物3与trxr作用1h后,可使trxr的活性分别降低至空白对照组水平的85%、57%和60%,并且,化合物2对trxr的抑制活性明显强于trxr抑制剂姜黄素。
[0135]
综上所述,本发明提供的新型荜茇酰胺类衍生物对硫氧还蛋白还原酶有很好的抑制作用,在抗肿瘤生物活性体外筛选实验中,该类衍生物对肿瘤细胞的生长也出现了一定的抑制作用,同时该类衍生物在a549人肺癌细胞中表现出促进活性氧生成的作用,因此,该类荜茇酰胺类衍生物可用于制备靶向硫氧还蛋白还原酶的促氧化抗癌药物。
[0136]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1