一种半芳香族聚酰胺树脂及其制备方法与流程
1.本发明属于高分子材料合成技术领域,具体涉及半芳香族聚酰胺树脂。
背景技术:
2.聚酰胺俗称尼龙,英文名称polyamide,是分子主链上含有重复酰胺基团—[nhco]—的热塑性树脂的总称,包括脂肪族聚酰胺、半芳香族聚酰胺和全芳香族聚酰胺。其中半芳香族聚酰胺在提供全芳香族聚酰胺刚性和耐热性的同时,又保持了脂肪族聚酰胺的加工性能,而且具有较低的吸水率、较好的尺寸稳定性、较高的强度和模量,在汽车内燃机部件、耐热电器部件、传动部件和表面贴装技术(smt)等领域得到越来越广泛的应用。
[0003]
与脂肪族聚酰胺产品类似,半芳香族聚酰胺产品也是通过二元酸和二元胺和/或内酰胺缩聚得到,但由于其含有芳香族二元酸或芳香族二元胺,形成的酰胺盐熔点高、在水溶液中溶解度低,在缩聚保压排出水汽的过程中,酰胺盐或低聚物常常会析出结块,导致反应釜无法搅拌、反应体系均一性变差、体系配比失衡、粘度无法控制、出现气泡甚至暴聚现象,降低了产成品率,甚至直接导致生产设备损坏及报废,极大干扰了半芳香族聚酰胺产品的顺利生产和产品品质的均一性。
[0004]
最直接的解决办法是提高生产设备的压力等级,使得反应体系在较高的压力下保压排出水汽,保证体系中有足够的水含量溶解芳香族酰胺盐及低聚物,但该方法需要增加生产设备的厚度、增加设备投资,而且体系中水含量增加,降低了反应速率和聚合度、耐热性能,增加了体系在高反应温度下发生副反应的概率,不利于产品品质的提升,同时降低了生产效率。而且升高反应压力等级会大大提升生产过程的安全风险、加重出现生产事故后破坏的严重程度。
[0005]
另外,还可以通过减少半芳香族聚酰胺产品中芳香单元的加入量,从而降低保压排气过程中芳香族酰胺盐结块的风险,但减少芳香单元,同样会降低最终产品的耐温性和模量等力学性能,无法满足客户的使用要求。
[0006]
除此之外,很多文献资料经常采用先在较低的温度和压力下聚合得到分子量较小的低聚物、然后采用后聚合或固相缩聚继续聚合的方法,得到所需分子量的半芳香族聚酰胺产品。该方法可以有效解决聚合设备压力等级高、产品均一性差的问题,同时保证了产品的芳香单元含量、耐温性和模量等性能。但该方法增加了设备数量、延长了反应过程,增加了反应工序和生产成本,不利于产品的市场竞争力。而且固相缩聚后的产品为粉末,需要经过熔融造粒才能转化成可销售的切片,继续增加了生产工序和成本,增加的熔融造粒过程还会导致产品品质一定程度上的下降。
[0007]
因此,还有必要对半芳香族聚酰胺产品及其生产工艺进行进一步优化。
技术实现要素:
[0008]
本发明的目的在于克服现有技术的不足之处,提供了一种半芳香族聚酰胺树脂及其制备方法。
[0009]
本发明的目的之一在于制备一种新型的半芳香族聚酰胺树脂,本发明通过在二元酸和二元胺和/或内酰胺缩聚的过程中,加入有机溶剂改善半芳香族酰胺盐及低聚物在体系中的溶解度,减小了半芳香族酰胺盐及低聚物发生析出结块的风险,保证了反应体系均一性、体系配比平衡和体系粘度可控性,从而有利于半芳香族聚酰胺产品的顺利生产和产品品质的均一性。同时降低了聚合设备压力等级,减少了设备投资和生产的安全风险。一步法缩聚反应缩短了生产流程和工序,降低了生产成本,增加了市场竞争力,而且所得产品是可直接销售的切片,避免了二次造粒对产品品质的影响。加入的有机溶剂在后期泄压过程排出循环使用,不会对产品品质和环境产生不利影响。
[0010]
本发明的另一目的是提供这种半芳香族聚酰胺树脂的制备方法。
[0011]
为实现上述目的,本发明采用的技术方案之一是:
[0012]
一种半芳香族聚酰胺树脂,所述半芳香族聚酰胺树脂的原料按重量份数计,包括:二元酸20~70份,二元胺20~50份,氨基酸或内酰胺0~30份,封端剂0.05~1份,催化剂0.01~0.5份,水30~160份,对酰胺盐的溶解度>0.001g的有机溶剂5~50份。
[0013]
优选地,所述二元酸为30~65份;
[0014]
优选地,所述二元胺为20~45份;
[0015]
优选地,所述氨基酸或内酰胺为0~20份;
[0016]
优选地,所述封端剂为0.1~0.5份;
[0017]
优选地,所述催化剂为0.02~0.2份;
[0018]
优选地,所述水为40~150份;
[0019]
优选地,所述有机溶剂为5~25份。
[0020]
其中,所述的二元酸选自芳香族二元酸、脂肪族二元酸、脂环族二元酸中的一种或几种的混合物。
[0021]
所述的芳香族二元酸选自对苯二甲酸、间苯二甲酸、5
‑
磺酸钠间苯二甲酸、5
‑
羟基间苯二甲酸、邻苯二甲酸、2
‑
甲基对苯二甲酸、5
‑
叔丁基间苯二甲酸、萘二甲酸中的一种或几种的混合物,进一步优选对苯二甲酸、间苯二甲酸中的一种或两种的混合物。
[0022]
所述的脂肪族二元酸选自直连或带支链的脂肪族c2~c36的二元酸,优选草酸、丙二酸、二甲基丙二酸、琥珀酸、3,3
‑
二乙基琥珀酸、戊二酸、2,2
‑
二甲基戊二酸、己二酸、2
‑
甲基己二酸、2,4,4
‑
三甲基己二酸、庚二酸、辛二酸、壬二酸、癸二酸、十一烷二酸、十二烷二酸、十三烷二酸、十四烷二酸、十五烷二酸、十六烷二酸、十八烷二酸、十八烯二酸、二十烷二酸、二十二烷二酸中的一种或几种的混合物,进一步优选自琥珀酸、戊二酸、己二酸、辛二酸、癸二酸、十一烷二酸、十二烷二酸中的一种或几种的混合物。
[0023]
所述的脂环族二元酸选自带脂环的c6~c36的二元酸,优选1,3
‑
环戊二甲酸、1,2
‑
环己烷二甲酸、4
‑
甲基
‑
1,2
‑
环己烷二甲酸、顺式/反式
‑
1,3
‑
环己烷二甲酸、顺式/反式
‑
1,4
‑
环己烷二甲酸、环庚烷二甲酸、环辛烷二甲酸、环癸烷二甲酸、1,3
‑
环己烷二乙酸、1,4
‑
环己烷二乙酸中的一种或几种的混合物,进一步优选顺式/反式
‑
1,4
‑
环己烷二甲酸。
[0024]
所述的二元胺选自脂肪族二元胺、芳香族二元胺、脂环族二元胺中的一种或几种的混合物。二元胺和反应体系中二元酸的量基本等摩尔量,添加量过多或过少都会导致产品粘度降低、力学性能变差、不具备实用价值。
[0025]
所述的脂肪族二元胺选自乙二胺、1
‑
丁基
‑
乙二胺、丙二胺、1,2
‑
丙二胺、丁二胺、
1,3
‑
丁二胺、1,1
‑
二甲基丁二胺、1,2
‑
二甲基丁二胺、1,3
‑
二甲基丁二胺、1,4
‑
二甲基丁二胺、2,3
‑
二甲基丁二胺、1
‑
乙基丁二胺、2
‑
乙基丁二胺、戊二胺、1,3
‑
戊二胺、2
‑
甲基
‑
1,5
‑
戊二胺、3
‑
甲基
‑
1,5
‑
戊二胺、2,2
‑
二甲基戊二胺、2
‑
丁基
‑2‑
乙基
‑
1,5
‑
戊二胺、己二胺、2
‑
甲基己二胺、3
‑
甲基己二胺、1
‑
丁基己二胺、2,2
‑
二甲基己二胺、2,4
‑
二甲基己二胺、2,5
‑
二甲基己二胺、3,3
‑
二甲基己二胺、2,4
‑
二乙基己二胺、2,2,4
‑
三甲基己二胺、2,4,4
‑
三甲基己二胺、庚二胺、2,2
‑
二甲基庚二胺、2,3
‑
二甲基庚二胺、2,4
‑
二甲基庚二胺、2,5
‑
二甲基庚二胺、辛二胺、2
‑
甲基
‑
1,8
‑
辛二胺、3
‑
甲基
‑
1,8
‑
辛二胺、4
‑
甲基
‑
1,8
‑
辛二胺、1,3
‑
二甲基辛二胺、1,4
‑
二甲基辛二胺、2,2
‑
二甲基辛二胺、2,4
‑
二甲基辛二胺、3,3
‑
二甲基辛二胺、3,4
‑
二甲基辛二胺、4,4
‑
二甲基辛二胺、4,5
‑
二甲基辛二胺、2,2,7,7
‑
四甲基辛二胺、壬二胺、5
‑
甲基壬二胺、癸二胺、十一烷二胺、十二烷二胺、十三烷二胺、十四烷二胺、十五烷二胺、十六烷二胺、十七烷二胺、十八烷二胺、十八烯二胺、十九烷二胺、二十烷二胺、二十二烷二胺、聚醚(peg、ppg、ptmg)二胺中的一种或几种的混合物,进一步优选自丁二胺、戊二胺、2
‑
甲基戊二胺、己二胺、辛二胺、壬二胺、癸二胺、十二烷二胺中的一种或几种的混合物。
[0026]
所述的氨基酸选自4
‑
氨基丁酸、6
‑
氨基己酸、7
‑
氨基庚酸、8
‑
氨基辛酸、9
‑
氨基壬酸、10
‑
氨基癸酸、11
‑
氨基十一烷酸、12
‑
氨基十二烷酸、4
‑
氨基环己烷羧酸、4
‑
(氨甲基)
‑
环己烷羧酸、对氨基苯甲酸、羟基色氨酸中的一种或几种的混合物,优选6
‑
氨基己酸、11
‑
氨基十一烷酸、12
‑
氨基十二烷酸中的一种或几种的混合物。
[0027]
所述的内酰胺选自β
‑
丙内酰胺、γ
‑
丁内酰胺、δ
‑
戊内酰胺、ε
‑
己内酰胺、庚内酰胺、辛内酰胺、壬内酰胺、癸内酰胺、十一内酰胺、月桂内酰胺中的一种或几种的混合物,优选ε
‑
己内酰胺、十一内酰胺、月桂内酰胺中的一种或几种的混合物。氨基酸和内酰胺可以任意比例加入反应体系中。
[0028]
所述的封端剂选自可以与聚酰胺分子链末端的氨基或羧基反应的单官能团化合物,可以是单羧酸、单胺、酸酐、单异氰酸酯、单酰氯、单酯、单醇等,优选乙酸、己酸、苯甲酸、乙胺、己胺、苯胺中的一种或几种的混合物。封端剂的作用是调节产品分子量,尤其是一元酸组分可以封住聚酰胺分子链末端的氨基,聚合时分子量分布变窄、催化剂变质减少,成型过程中瓦斯气体减少、脱模性能改善,防止在加工和使用过程中受热状态的热降解和氧化降解导致的性能恶化、变色;封端剂含量过高会导致产品粘度下降、分子量降低、力学性能变差;含量过低时末端官能团含量过高,导致熔融滞留时的凝胶化或劣化,而且在使用环境下产生着色或水解等问题。
[0029]
所述的催化剂选自磷酸、亚磷酸、次磷酸或其金属盐或酯,金属盐优选钾、钠、镁、钒、钙、锌、钴、锰、锡、钨、锗、钛、锑、镍等,酯优选甲酯、乙酯、异丙酯、丁酯、己酯、癸酯、异癸酯、十八烷酯、苯酯等。催化剂在体系中具有催化作用,加快反应速率,同时使主链中的分支含量降低、从而有助于减小聚酰胺产品的pdi;优选0.02~0.05份,用量过少,没有催化效果,用量过多、聚合度过大,难以加工。
[0030]
所述的有机溶剂选自对酰胺盐的溶解度>0.001g的有机溶剂,优选沸点>100℃且对酰胺盐的溶解度>0.001g的有机溶剂,进一步优选100℃<沸点<320℃且对酰胺盐的溶解度>0.001g的有机溶剂,更优选1,4
‑
二氧六环、甲苯、硝基乙烷、吡啶、4
‑
甲基
‑2‑
戊酮、辛烷、吗啉、氯苯、对二甲苯、间二甲苯、邻二甲苯、n,n
‑
二甲基甲酰胺、环己酮、n,n
‑
二甲基乙酰胺、糠醛、n
‑
甲基甲酰胺、二甲基亚砜、n
‑
甲基吡咯烷酮、甲酰胺、硝基苯、乙酰胺、六甲
基磷酸三酰胺、喹啉、丁二腈、环丁砜中的一种或几种的混合物,最优选1,4
‑
二氧六环、甲苯、吡啶、辛烷、对二甲苯、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二甲基亚砜、n
‑
甲基吡咯烷酮中的一种或几种的混合物。对酰胺盐的溶解度过低,在体系中起不到促进酰胺盐的溶解、防止其析出结块的作用。沸点过低,在保压过程中先于水分排出,无法在保压后期起到增加酰胺盐溶解度的作用,沸点过高,在排气末端都未能从反应体系排出,残留在反应产物中影响最终产品性能。有机溶剂优选5~25份,添加量过少,对酰胺盐及低聚物的溶解效果不佳,不利于防止其析出结块,添加量过多后期排出费时费能,降低生产效率,增加生产成本。
[0031]
在一个优选的实施例中,所述二元酸为30~35份;所述二元胺为20~35份;所述封端剂为0.1~0.25份;所述催化剂为0.03~0.05份;所述水为40~45份;所述有机溶剂为6~14份;所述二元酸为对苯二甲酸、间苯二甲酸或己二酸中的至少一种;所述二元胺为己二胺、壬二胺或癸二胺中的至少一种;所述封端剂为乙酸或苯甲酸中的至少一种;所述有机溶剂为n,n
‑
二甲基甲酰胺、n
‑
甲基吡咯烷酮或甲苯中的至少一种。
[0032]
为实现上述目的,本发明采用的技术方案之二是:
[0033]
一种半芳香族聚酰胺树脂的制备方法,将所述各原料置于微正压条件的保护气体中,搅拌并升温至60~150℃,恒温反应1~3h,之后继续升温至200~240℃,使压力达到1.5~3.0mpa,保温1~5h;继续升温并维持恒压状态;升温至240~340℃后,经过0.5~2h泄压至0mpa,之后在常压恒温反应0.1~1h,得到产物。
[0034]
其中,所述微正压指的是不高于0.06mpa,例如为0~0.05mpa。所述保护气体例如为氮气。
[0035]
具体地,所述制备方法包括以下步骤:
[0036]
(1)称取重量份为20~70份的二元酸、20~50份二元胺、0~30份氨基酸/内酰胺、0.05~1份封端剂、0.01~0.5份催化剂、30~160份去离子水、10~50份有机溶剂加入到高压反应釜内,高压反应釜内抽真空、充氮气,重复三次除去反应釜内残留的空气,置换完成后高压反应釜保留微正压0.05mpa;
[0037]
(2)在100r/min的搅拌条件下高压反应釜加热升温至60~150℃,恒温反应1~3h,之后继续升温至200~240℃,压力达到1.5~3.0mpa,保温1~5h;
[0038]
(3)继续升温,同时通过释放高压反应釜内水蒸气和有机溶剂的方法使该高压反应釜处于恒压状态,升温至240~340℃时,经过0.5~2h将釜内压力缓慢泄压至0mpa,之后在常压恒温反应0.1~1h;
[0039]
(4)将聚合物从高压反应釜经过模头挤出、水槽冷却、切粒,得到新型聚酰胺产品。
[0040]
本发明所涉及的设备、试剂、工艺、参数等,除有特别说明外,均为常规设备、试剂、工艺、参数等,不再作实施例。
[0041]
本发明所列举的所有范围包括该范围内的所有点值。
[0042]
本发明同现有技术相比,具有如下优点:
[0043]
1.本发明通过在半芳香族聚酰胺树脂聚合过程中加入有机溶剂,增加酰胺盐及低聚物的溶解度,从而降低了酰胺盐及低聚物在水中完全溶解所需的温度及压力,降低了对设备的压力要求,节约了设备投资和生产成本,增强了产品的市场竞争力。
[0044]
2.本发明通过加入有机溶剂,降低了聚合设备的压力等级,减小了生产过程的安
全风险和出现生产事故后破坏的严重程度。
[0045]
3.本发明通过加入有机溶剂,促进了酰胺盐和低聚物的溶解度,防止在水汽排出过程中酰胺盐析出、反应体系出现固相分离、均一性变差的问题出现,有利于半芳香族聚酰胺产品的顺利生产和产品品质的均一性。
[0046]
4.本发明通过一步法合成半芳香族聚酰胺产品,减少了设备数量和反应工序,提高了生产效率,降低了生产成本,有利于增加产品的市场竞争力。
[0047]
5.本发明通过一步法合成半芳香族聚酰胺产品,所得产品是可以直接销售的切片,避免了采用固相缩聚工艺需要二次造粒,对产品品质的影响。
[0048]
6.本发明加入的有机溶剂,在后期较高温度泄压过程中会随水汽一块排出,不会残留在产品体系中、对产品品质造成影响。
[0049]
7.本发明加入的有机溶剂,在后期较高温度泄压过程中排出后通过冷凝、再次进入下次反应过程中循环使用,不会对环境产生影响。
[0050]
8.本发明通过加入少量的有机溶剂,跟常规全部使用有机溶剂的溶液聚合有本质区别,本质还是属于熔融缩聚。同时减少了有机溶剂的使用量,也减少了回收有机溶剂的工作量,降低生产成本。
具体实施方式
[0051]
下面通过实施例具体说明本发明的内容:
[0052]
在以下提供的实施例中,采用下面检测方法:
[0053]
熔点t
m
和玻璃化转变温度t
g
:根据iso11357:用差示扫描量热仪(梅特勒
‑
托利多公司的dsc3)以20℃/min的速度升温至350℃、停留2min、再以20℃/min的速度降温至25℃、再停留2min、再以20℃/min的速度升温至350℃,第二次升温曲线吸热峰值对应的温度即为熔点t
m
、半台阶高度法测试的玻璃化转变时的中点即为玻璃化转变温度t
g
。
[0054]
实施例1
[0055]
(1)称取1661g(10mol)对苯二甲酸、1723g(10mol)癸二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水、400g的n,n
‑
二甲基甲酰胺加入到高压反应釜内,高压反应釜内抽真空、充氮气,重复三次除去反应釜内残留的空气,置换完成后高压反应釜保留微正压0.05mpa;
[0056]
(2)在100r/min的搅拌条件下高压反应釜加热升温至130℃,恒温反应2h,之后继续升温至220℃,压力达到2.0mpa,保温2h;
[0057]
(3)继续升温,同时通过释放高压反应釜内水蒸气和n,n
‑
二甲基甲酰胺的方法使该高压反应釜处于2.0mpa的恒压状态,升温至320℃时,经过1h将釜内压力缓慢泄压至0mpa,之后在常压恒温反应0.3h;
[0058]
(4)将聚合物从高压反应釜经过模头挤出、水槽冷却、切粒,得到聚对苯二甲酰癸二胺树脂。实施例1的原料单体及其性能列于表1中。
[0059]
实施例2
[0060]
(1)称取1163g(7mol)对苯二甲酸、498g(3mol)间苯二甲酸、1162g(10mol)己二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水、400g的n
‑
甲基吡咯烷酮加入到高压反应釜内,高压反应釜内抽真空、充氮气,重复三次除去反应釜内残留的空气,置
换完成后高压反应釜保留微正压0.05mpa;
[0061]
(2)在100r/min的搅拌条件下高压反应釜加热升温至100℃,恒温反应1.5h,之后继续升温至228℃,压力达到2.5mpa,保温2.5h;
[0062]
(3)继续升温,同时通过释放高压反应釜内水蒸气和n
‑
甲基吡咯烷酮的方法使该高压反应釜处于2.5mpa的恒压状态,升温至330℃时,经过0.8h将釜内压力缓慢泄压至0mpa,之后在常压恒温反应0.2h;
[0063]
(4)将聚合物从高压反应釜经过模头挤出、水槽冷却、切粒,得到聚对苯二甲酰己二胺
‑
co
‑
间苯二甲酰己二胺共聚树脂。实施例2的原料单体及其性能列于表1中。
[0064]
实施例3
[0065]
(1)称取914g(5.5mol)对苯二甲酸、658g(4.5mol)己二酸、1162g(10mol)己二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水、500g的n,n
‑
二甲基甲酰胺加入到高压反应釜内,高压反应釜内抽真空、充氮气,重复三次除去反应釜内残留的空气,置换完成后高压反应釜保留微正压0.05mpa;
[0066]
(2)在100r/min的搅拌条件下高压反应釜加热升温至80℃,恒温反应2.5h,之后继续升温至205℃,压力达到1.8mpa,保温1.5h;
[0067]
(3)继续升温,同时通过释放高压反应釜内水蒸气和n,n
‑
二甲基甲酰胺的方法使该高压反应釜处于1.8mpa的恒压状态,升温至315℃时,经过1.5h将釜内压力缓慢泄压至0mpa,之后在常压恒温反应0.8h;
[0068]
(4)将聚合物从高压反应釜经过模头挤出、水槽冷却、切粒,得到聚对苯二甲酰己二胺
‑
co
‑
己二酰己二胺共聚树脂。实施例3的原料单体及其性能列于表1中。
[0069]
实施例4
[0070]
(1)1661g(10mol)对苯二甲酸、1583g(10mol)壬二胺、6g(0.1mol)封端剂醋酸、2g催化剂次磷酸钠、2000g去离子水、600g的甲苯加入到高压反应釜内,高压反应釜内抽真空、充氮气,重复三次除去反应釜内残留的空气,置换完成后高压反应釜保留微正压0.05mpa;
[0071]
(2)在100r/min的搅拌条件下高压反应釜加热升温至140℃,恒温反应1h,之后继续升温至223℃,压力达到2.2mpa,保温3h;
[0072]
(3)继续升温,同时通过释放高压反应釜内水蒸气和甲苯的方法使该高压反应釜处于2.2mpa的恒压状态,升温至312℃时,经过0.6h将釜内压力缓慢泄压至0mpa,之后在常压恒温反应1h;
[0073]
(4)将聚合物从高压反应釜经过模头挤出、水槽冷却、切粒,得到聚对苯二甲酰壬二胺树脂。实施例4的原料单体及其性能列于表1中。
[0074]
对比例1
[0075]
称取1661g(10mol)对苯二甲酸、1723g(10mol)癸二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水加入到高压反应釜内,除此之外,其它与实施例1一样,在进行到保压(2.0mpa)升温阶段(265℃)时,10t盐和低聚物开始析出,阻碍反应釜搅拌,反应中止。对比例1的原料单体及对比结果列于表2中。
[0076]
对比例2
[0077]
称取1661g(10mol)对苯二甲酸、1723g(10mol)癸二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水加入到高压反应釜内,升温到235℃开始保压
(2.8mpa),除此之外,其它与实施例1一样,合成得到聚对苯二甲酰癸二胺树脂,对比例2的原料单体及其性能列于表2中。
[0078]
对比例3
[0079]
称取1163g(7mol)对苯二甲酸、498g(3mol)间苯二甲酸、1162g(10mol)己二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水加入到高压反应釜内,除此之外,其它与实施例2一样,在进行到保压(2.5mpa)升温阶段(261℃)时,6t盐和低聚物开始析出,阻碍反应釜搅拌,反应中止。对比例3的原料单体及对比结果列于表2中。
[0080]
对比例4
[0081]
称取1163g(7mol)对苯二甲酸、498g(3mol)间苯二甲酸、1162g(10mol)己二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水加入到高压反应釜内,升温到240℃开始保压(3.5mpa),除此之外,其它与实施例2一样,合成得到聚对苯二甲酰己二胺
‑
co
‑
间苯二甲酰己二胺共聚树脂,对比例4的原料单体及其性能列于表2中。
[0082]
对比例5
[0083]
称取914g(5.5mol)对苯二甲酸、658g(4.5mol)己二酸、1162g(10mol)己二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水加入到高压反应釜内,升温到205℃开始保压(1.8mpa),除此之外,其它与实施例3一样,在进行到保压(1.8mpa)升温阶段(263℃)时,6t盐和低聚物开始析出,阻碍反应釜搅拌,反应中止。对比例5的原料单体及对比结果列于表2中。
[0084]
对比例6
[0085]
称取914g(5.5mol)对苯二甲酸、658g(4.5mol)己二酸、1162g(10mol)己二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水、500g乙醇(沸点78℃)加入到高压反应釜内,升温到205℃开始保压(1.8mpa),除此之外,其它与实施例3一样,在进行到保压(1.8mpa)升温阶段(261℃)时,6t盐和低聚物开始析出,阻碍反应釜搅拌,反应中止。对比例6的原料单体及对比结果列于表2中。
[0086]
对比例7
[0087]
称取914g(5.5mol)对苯二甲酸、658g(4.5mol)己二酸、1162g(10mol)己二胺、12g(0.1mol)封端剂苯甲酸、2g催化剂次磷酸钠、2000g去离子水、500g氢化三联苯(沸点352.8℃)加入到高压反应釜内,除此之外,其它与实施例3一样,最后排出的产品处于溶胀状态,无法顺利拉条切粒。对比例7的原料单体及对比结果列于表2中。
[0088]
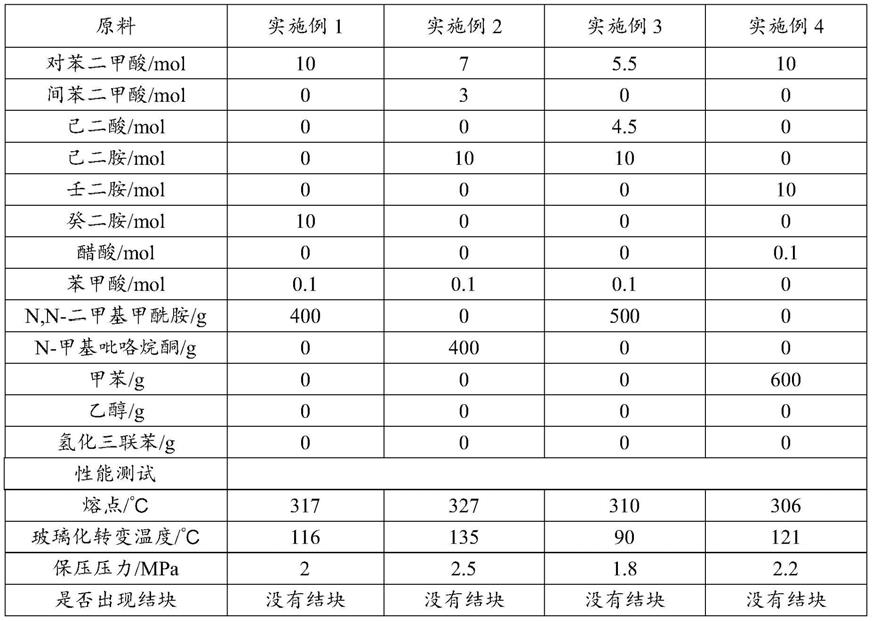
表1实施例的配方及性能测试表
[0089][0090]
表2对比例的配方及性能测试表
[0091][0092]
经过比较实施例1和对比例1的实验过程可知,实施例1在对比例1的基础上增加了400g的n,n
‑
二甲基甲酰胺,其余原料和聚合条件完全相同,实施例1在保压阶段10t盐和低聚物没有析出结块、可以顺利聚合,而对比例1在同样的保压压力下进行到265℃时,10t盐和低聚物开始析出,阻碍反应釜搅拌,反应中止。说明实施例1加入的400g的n,n
‑
二甲基甲酰胺确实可以起到增加10t盐和低聚物在反应体系中溶解度的作用,防止体系中水分减少
时10t盐和低聚物析出,使得聚合过程可顺利进行。通过对所得产品测试熔点t
m
和玻璃化转变温度t
g
,与之前文献报道和对比例2的产品性能基本相同,说明实施例1加入的n,n
‑
二甲基甲酰胺在产品中没有残留,不会影响最终产品的使用性能。
[0093]
对比例2在对比例1的基础上,将保压压力从2.0mpa增加到2.8mpa,其余原料和聚合条件完全相同,在保压阶段10t盐和低聚物没有析出结块、可以顺利聚合,说明增加保压压力、增加保压阶段反应体系中的水分确实可以防止10t盐和低聚物析出,使得聚合过程顺利进行。但是增加保压压力,就需要增大生产设备的耐压等级、增加设备的壁厚,会极大增加设备投资和生产成本,影响产品的市场竞争力。
[0094]
通过比较实施例2和对比例3、对比例4,以及实施例3和对比例5的实验过程可知,实施例2和实施例3加入有机溶剂在较低的压力下就可以顺利聚合,而同样原料和聚合条件的对比例3和对比例5在保压阶段就会出现6t盐和低聚物析出,反应中止的现象。而对比例4在实施例3的基础上升高保压压力,就可以避免6t盐和低聚物的析出,反应顺利进行。再次说明本发明实施例加入有机溶剂可以明显改善保压阶段酰胺盐和低聚物的溶解度,降低聚合设备的压力等级,节省设备投资和生产成本,增加产品市场竞争力,降低安全风险,保证反应体系及产品的均一性。
[0095]
实施例6加入低沸点的乙醇,在保压过程中先于水蒸气馏出,体系中剩余量很少,无法起到溶解半芳香族酰胺盐和低聚物的作用,产品在较低压力下出现结块、反应中止。实施例7加入高沸点的氢化三联苯,在保压及后续泄压过程中都无法顺利排出,导致产品一直处于溶胀状态,无法顺利拉条切粒,也无法顺利生产和进行后续的加工应用。测试熔点t
m
和玻璃化转变温度t
g
,由于聚合物中大量有机溶剂存在,起增塑剂的作用,与实施例3相比,熔点t
m
和玻璃化转变温度t
g
都明显降低。
[0096]
此外,本发明加入的有机溶剂不会残留在产品体系中、对产品品质造成影响,有机溶剂在后期较高温度泄压过程中排出后通过冷凝、再次进入下次反应过程中循环使用,不会对环境产生影响。
[0097]
上述对实施例的描述是为了便于该技术领域的普通技术人员能理解和使用本发明。熟悉本领域技术人员显然可以容易的对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中,而不必经过创造性的劳动。因此,本发明不限于上述实施例。本领域技术人员根据本发明的原理,不脱离本发明的范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1