一种制备含吡啶并环己烷手性氮磷配体L-8的方法与流程
一种制备含吡啶并环己烷手性氮磷配体l-8的方法
技术领域
[0001]
本发明属于医药中间体手性配体技术领域,具体涉及一种制备含吡啶基衍生的手性氮磷配体l-8的方法。
背景技术:
[0002]
手性氮磷配体是一类杂双齿配体,这类配体的结构可调性大,通过对取代基、配位点肯配位构型的改变,可以提高其金属络合物的催化活性,其应用前景十分广阔。
[0003]
含吡啶基衍生的手性氮磷配体l-8(结构如下),经过与金属铱络合后形成的配合物,在非官能化烯烃的不对称氢化上取得了非常优异的结果。
[0004][0005]
然而该类配体的合成步骤较为繁琐,需要对现有合成路线进行优化和改进,以便于能够克级以上规模的方便制备。
技术实现要素:
[0006]
本发明提供一种操作简便稳定、收率较高、步骤较短的合成含吡啶基衍生手性氮磷配体l-8的方法。从环酮出发,依次经过加成、环合、氯代、不对称硼化、氧化、偶联、成酯等步骤后完成得到手性氮磷l-8配体。该路线相对比较容易实现较大规模的制备,克服了传统路线中第一步关环反应和手性醇制备时收率低,通过选择合适的手性配体,与丁基锂结合,实现了不对称合成手性醇,避免了文献中采用手性分离柱方式。
[0007]
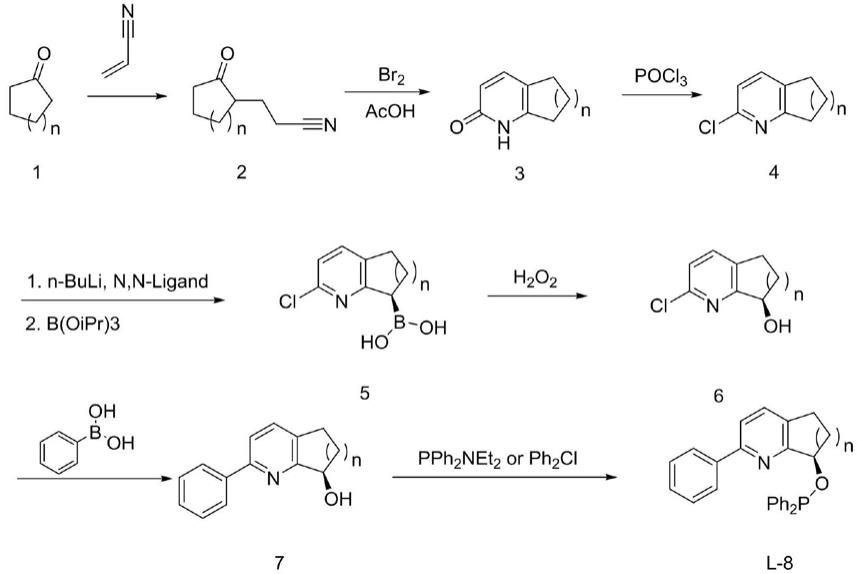
本发明所提供合成含吡啶基衍生手性氮磷配体l-8的方法,其反应方程式如下:
[0008][0009]
本发明的技术方案的实现,其特征在于,包括步骤如下:
[0010]
第一步:3-(2-氧代环基)丙腈2的合成
[0011]
将环酮1与四氢吡咯脱水生成烯胺,然后与丙烯腈加成反应得到3-(2-氧代环基)丙腈2。
[0012]
其中,反应溶剂为甲苯或1,4-二氧六环,反应温度选自20-110℃;环酮选自环戊酮、环己酮和环庚酮。环酮、四氢吡咯与丙烯腈摩尔比为1:1.1-1.4:1.0-1.1。
[0013]
第二步:1,5,6,7-四氢-2h-环[b]吡啶-2-酮3的合成
[0014]
3-(2-氧代环基)丙腈2在醋酸溶剂中,与溴素进行亲电加成,关环得到1,5,6,7-四氢-2h-环[b]吡啶-2-酮3。其中,反应温度选自0℃至30℃。
[0015]
第三步:2-氯-6,7-二氢-5h-环[b]吡啶4的合成
[0016]
1,5,6,7-四氢-2h-环[b]吡啶-2-酮3与三氯氧磷回流反应得到2-氯-6,7-二氢-5h-环[b]吡啶4。其中,三氯氧磷在反应中即做反应物,又做为溶剂,反应温度选自80℃至105℃。
[0017]
第四步:r-(2-氯-6,7-二氢-5h-环[b]吡啶)硼酸5的合成
[0018]
2-氯-6,7-二氢-5h-环[b]吡啶4与(1s,2s)-双二甲基-(3,3-二甲基丁基)环己烷-1,2-二胺混合,正丁基锂拔氢再与硼酸三异丙酯反应得到r-(2-氯-6,7-二氢-5h-环戊[b]吡啶)硼酸5。
[0019]
其中,反应溶剂选自四氢呋喃、2-甲基四氢呋喃、二乙氧基甲烷等;2-氯-6,7-二氢-5h-环[b]吡啶4、(1s,2s)-双二甲基-(3,3-二甲基丁基)环己烷-1,2-二胺、正丁基锂与硼酸三异丙酯摩尔比为1:1-1.4:1-1.4:1.1-2.0。
[0020]
第五步:r-2-氯-6,7-二氢-5h-环[b]吡啶-7-醇6的合成
[0021]
r-(2-氯-6,7-二氢-5h-环[b]吡啶)硼酸5在氢氧化钠水溶液中与双氧水反应得到r-2-氯-6,7-二氢-5h-环[b]吡啶-7-醇6。
[0022]
其中,反应温度选自5℃至25℃。r-(2-氯-6,7-二氢-5h-环[b]吡啶)硼酸与双氧水、氢氧化钠的摩尔比例为1:3-11:9.5-9.8。
[0023]
第六步:r-2-苯基-6,7-二氢-5h-环[b]吡啶-7-醇7的合成
[0024]
r-2-氯-6,7-二氢-5h-环[b]吡啶-7-醇6与苯硼酸和无机碱在钯催化剂存在下,发生偶联得到r-2-苯基-6,7-二氢-5h-环[b]吡啶-7-醇7。
[0025]
其中,反应温度选自80℃至110℃;钯催化剂选自pd(pph3)4、pdcl2(pph3)2或pdcl2dppf;无机碱选自碳酸钾或碳酸钠水溶液。r-2-氯-6,7-二氢-5h-环戊[b]吡啶-7-醇6、钯催化剂、苯硼酸和醋酸钾的摩尔比例为1:0.005-0.02:0.95-1.1:1.5-2。
[0026]
第七步:r-7-[(二苯基膦酰基)氧基]-2-苯基-6,7-二氢-5h-环[b]吡啶8的合成
[0027]
r-2-苯基-6,7-二氢-5h-环[b]吡啶-7-醇7与二乙胺基二苯基磷、4,5-二氯咪唑、有机胺或者与去质子试剂和二苯基氯化磷合成得到r-7-[(二苯基膦酰基)氧基]-2-苯基-6,7-二氢-5h-环[b]吡啶8。
[0028]
其中,反应温度采用前者为在1,2-二氯乙烷回流温度下进行,采用后者为-10℃至20℃下进行。有机胺选自三乙胺或二异丙基乙基胺。去质子试剂选自氢化钠或正丁基锂。
[0029]
前者方法中:r-2-苯基-6,7-二氢-5h-环[b]吡啶-7-醇7、二乙胺基二苯基磷、4,5-二氯咪唑与有机胺摩尔比为1:1.0-1.3:2.0-2.5:1.1-1.3。
[0030]
后者方法中:r-2-苯基-6,7-二氢-5h-环[b]吡啶-7-醇、去质子试剂与二苯基氯化磷摩尔比为1:1.0-1.3:1.1-1.5。
[0031]
进一步地,得到的配体l-8包括如下三种结构:
[0032][0033]
发明有益效果:
[0034]
本发明采用方法,相比文献路线,反应更加合理有效,原料市场上均可以直接获取,每步反应收率高,为该类化合物克级以上制备最有有效的方法。两种构型的原料均可通过第四步中,采用不同构型的配体来进行切换。
具体实施方式
[0035]
本发明以下实施例中所述的室温均值20-25℃。除非特别指出,所述的试剂不特别说明均为不经纯化直接使用。所有溶剂均购自商业化供应商,并且不经处理就可使用。反应通过tlc、gc、hplc分析,通过起始材料的消耗来判断反应的终止。其中,n=1,2,3分别标记为对应的a,b,c.
[0036]
实施例1
[0037]
第一步:3-(2-氧代环戊基)丙腈2a的合成
6,7-二氢-5h-环戊[b]吡啶1.29g,hplc:99.1%,收率84%。1hnmr(400mhz,cdcl3):2.18-1.95(m,2h),2.64-2.53(m,2h),2.84-2.71(m,2h),6.97(d,j=8.1hz,1h),7.21(d,j=7.9hz,1h).ms[m+h]
+
=153.6.
[0050]
采用相同的方法,得到产物4b和4c收率分别为88%和81%。
[0051]
实施例4
[0052]
第四步:(r)-(2-氯-6,7-二氢-5h-环戊[b]吡啶)硼酸5a的合成。
[0053][0054]
将2-氯-6,7-二氢-5h-环戊[b]吡啶4a(15.3,0.1mol)、四氢呋喃55ml和(1s,2s)-双二甲基-(3,3-二甲基丁基)环己烷-1,2-二胺(37g,1.2eq),控制温度在-75℃至-65℃滴加2.5m正丁基锂1.2eq,滴毕保温2小时后,再滴加含硼酸三异丙酯(37.6g,2eq)的四氢呋喃溶液,控制温度在-75℃至-65℃保温1小时,缓慢升至-10℃,滴加100ml水进行淬灭,再滴加冰醋酸,调节ph=5~6。减压浓缩除去四氢呋喃,每次加入乙酸乙酯50ml进行萃取,共萃取3次。合并有机相,饱和碳酸氢钠水洗,氯化钠水溶液洗,浓缩至不流液,二氯甲烷和正庚烷进行重结晶得到(r)-(2-氯-6,7-二氢-5h-环戊[b]吡啶)硼酸12.2g,hplc:98.6%,97%ee,收率62%;1hnmr(400mhz,dmso-d6):1.92-1.83(m,1h),1.95-2.08(m,1h),2.51-2.45,(m,1h),2.64-2.58(m,1h),3.62(br,1h),7.12(d,j=8.1hz,1h),7.68(d,j=7.9hz,1h).9.2(t,2h).ms[m+h]
+
=197.43.
[0055]
其中,(1s,2s)-双二甲基-(3,3-二甲基丁基)环己烷-1,2-二胺,cas,767291-67-8,化学结构式为
[0056]
水相用10%氢氧化钠水溶液调节ph=12-13,二氯甲烷萃取,浓缩回收(1s,2s)-双二甲基-(3,3-二甲基丁基)环己烷-1,2-二胺。
[0057]
采用相同的方法,得到产物5b和5c收率/对映选择性分别为73%/98%ee和59%/99%ee。
[0058]
实施例5
[0059]
第五步:(r)-2-氯-6,7-二氢-5h-环戊[b]吡啶-7-醇6a的合成
[0060][0061]
将(r)-(2-氯-6,7-二氢-5h-环戊[b]吡啶)硼酸(4.2g,0.02mol)、2%氢氧化钠水溶液(135g)和四氢呋喃(150ml),控制温度在0-5℃滴加28%双氧水(0.2mol,10eq)。室温搅拌过夜,hplc检验原料<0.3%,减压蒸馏除去四氢呋喃,加入4mol/l盐酸,调节ph=2-3,每次用乙酸乙酯30ml萃取杂质,共萃取3次。水相用饱和碳酸钠调节ph=8-9,乙酸乙酯萃取,合并有机相,浓缩后得到(r)-2-氯-6,7-二氢-5h-环戊[b]吡啶-7-醇3.3g,hplc97.3%,98.6%ee,收率91%。1h nmr(400mhz,cdc13):7.53(d,j=8.0hz,1h),7.18(d,j=8.0hz,1h),5.32-5.09(m,1h),3.56(br,1h),3.11-2.93(m,1h),2.88-2.71(m,1h),2.65-2.48(m,1h),2.17-2.02(m,1h).ms[m+h]+=169.61.产物绝对构型通过与文献旋光符号比较,确定为r型。
[0062]
第五步:参照实施例1第五步的操作,其中n=1,2,3
[0063]
实验编号n反应温度收率1n=10℃至25℃91%2n=20℃至25℃89%3n=30℃至25℃88%
[0064]
备注:n=1,原料为环戊酮;n=2,原料为环己酮;n=3,原料为环庚酮。n=2,1h nmr(400mhz,cdcl3)δ:7.45(d,j=8.4hz,1h),7.15(d,j=8.0hz,1h),5.55-5.39(m,1h),4.55(t,j=4.8hz,1h),2.76-2.59(m,2h),1.95-1.80(m,3h),1.75-1.62(m,1h).ms[m+h]+=183.63.
[0065]
n=3,1h nmr(400mhz,cdcl3)δ:1.49-1.88(m,6h),2.85(d,j=8.1hz,2h),4.92(t,1h),5.68-5.59(m,1h),6.59(d,j=7.4hz,1h),6.21(d,j=7.2hz,1h),8.23(t,2h).ms[m+h]+=197.66.
[0066]
实施例6
[0067]
第六步:(r)-2-苯基-6,7-二氢-5h-环戊[b]吡啶-7-醇(7)的合成。
[0068][0069]
在氮气保护下,将(r)-2-氯-6,7-二氢-5h-环戊[b]吡啶-7-醇(9.2g,0.05mol)、双苯基磷二氯化钯(1.0%mol)、醋酸钾(7.35g,1.5eq)、苯硼酸(6.7g,1.1eq)和1,4-二氧六环(45ml)缓慢升温至70~75℃,保温反应1小时,hplc检测原料剩余<1%,减压蒸馏除去1,4-二氧六环,加入甲基叔丁基醚(125ml),过滤,滤饼用甲基叔丁基醚(25ml)淋洗,滤液浓缩
干,柱层析(乙酸乙酯/正庚烷=1:1)进行洗脱,得到(r)-2-苯基-6,7-二氢-5h-环戊[b]吡啶-7-醇6.7g,hplc 95.3%,收率64%。1h nmr(400mhz,cdcl3):7.96(d,j=8.8hz,2h),7.62(d,j=8.0hz,1h),7.36-7.58(m,4h),5.25(t,1h),3.26(br,1h),3.11-2.93(m,1h),2.87-2.81(m,1h),2.58-2.51(m,1h),2.07-2.02(m,1h).ms[m+h]+=211.26.
[0070]
第六步:参照实施例1第六步的操作,其中n=1,2,3
[0071]
实验编号n反应温度收率1n=125℃至70℃64%2n=225℃至75℃73%3n=325℃至75℃81%
[0072]
备注:n=1,原料为环戊酮;n=2,原料为环己酮;n=3,原料为环庚酮
[0073]
n=2,1hnmr(400mhz,cdcl3):1.74-1.87(m,2h),1.96-2.04(m,1h),2.31-2.38(m,1h),2.76-2.87(m,1h),4.44(br s,1h),4.72(t,j=6.0hz,1h),7.36-7.46(m,4h),7.53(d,j=8.0hz,1h),7.96(d,j=8.8hz,2h).ms[m+h]+=225.29.
[0074]
n=3,1hnmr(400mhz,cdcl3):7.96(d,j=8.8hz,2h),7.62(d,j=8.0,hz,1h),7.36-7.58(m,4h),5.68-5.59(m,1h),4.92(t,1h),2.85(d,j=8.1hz,2h),1.49-1.88(m,6h).ms[m+h]+=239.31
[0075]
实施例7
[0076]
第七步:(r)-7-[(二苯基膦酰基)氧基]-2-苯基-6,7-二氢-5h-环戊[b]吡啶及衍生物(8)的合成。
[0077][0078]
方法一、在氮气保护下,将(r)-2-苯基-6,7-二氢-5h-环戊[b]吡啶-7-醇(2.1g,0.01mol)、二乙氨基二苯基膦(3.0g,1.2eq)、4,5-二氯咪唑(2.7g,2eq)和三乙胺(1.2g,1.2eq)和1,2-二氯乙烷(50ml)室温下反应过夜,tlc检测反应完毕,反应液直接浓缩,快速柱层析(乙酸乙酯:正庚烷=1:5)洗脱,得到(r)-7-[(二苯基膦酰基)氧基]-2-苯基-6,7-二氢-5h-环戊[b]吡啶3.2g,hplc98.1%,99.1%ee,收率82%。
[0079]
n=1,1hnmr(400mhz,cdcl3):7.96(d,j=8.8hz,2h),7.62(d,j=8.0,hz,1h),7.46-7.58(m,4h),7.42(dd,6h),7.15(dd,4h),5.25(t,1h),3.26(br,1h),3.11-2.93(m,1h),2.87-2.81(m,1h),2.58-2.51(m,1h),2.07-2.02(m,1h).ms[m+h]+=395.43.
[0080]
n=2,1hnmr(400mhz,cdcl3):1.74-1.87(m,2h),1.96-2.04(m,1h),2.31-2.38(m,1h),2.76-2.87(m,1h),4.44(br,1h),4.72(t,j=6.0hz,1h),7.15(dd,4h),7.42(dd,6h),7.43-7.48(m,4h),7.53(d,j=8.0hz,1h),7.96(d,j=8.8hz,2h).ms[m+h]+=409.46.
[0081]
n=3,1hnmr(400mhz,cdcl3):7.96(d,j=8.8hz,2h),7.62(d,j=8.0,hz,1h),7.53-7.48(m,4h),7.42(dd,6h),7.15(dd,4h),5.68-5.59(m,1h),4.92(t,1h),2.85(d,j=8.1hz,2h),1.49-1.88(m,6h).ms[m+h]+=423.49.
[0082]
第七步:参照实施例7方法一的操作,其中n=1,2,3
[0083]
实验编号n反应温度收率1n=10℃至25℃82%2n=20℃至25℃81%3n=30℃至25℃77%
[0084]
方法二、在氮气保护下,将(r)-2-苯基-5,6,7,8-四氢喹啉-8-醇(2.3g,0.01mol)和四氢呋喃30ml室温下搅拌均匀,分批加入氢化钠(1.1eq),25-50℃反应1小时。冰浴下滴加含二苯基氯化磷(2.4g,1.1eq)的四氢呋喃(10ml)溶液。滴毕,升至室温反应8小时,tlc检测反应完毕,冰浴下加入5ml甲醇,硅藻土过滤,旋干溶剂中,直接快速柱层析得到(r)-8[(二苯基膦酰基)氧基]-2-苯基-5,6,7,8-四氢喹啉3.6g,hplc98.3%,ee98.7%,收率89%。
[0085]
n=1,1hnmr(400mhz,cdcl3):7.96(d,j=8.8hz,2h),7.62(d,j=8.0,hz,1h),7.46-7.58(m,4h),7.42(dd,6h),7.15(dd,4h),5.25(t,1h),3.26(br,1h),3.11-2.93(m,1h),2.87-2.81(m,1h),2.58-2.51(m,1h),2.07-2.02(m,1h).ms[m+h]+=395.43.
[0086]
n=2,1hnmr(400mhz,cdcl3):1.74-1.87(m,2h),1.96-2.04(m,1h),2.31-2.38(m,1h),2.76-2.87(m,1h),4.44(br,1h),4.72(t,j=6.0hz,1h),7.15(dd,4h),7.42(dd,6h),7.43-7.48(m,4h),7.53(d,j=8.0hz,1h),7.96(d,j=8.8hz,2h).ms[m+h]+=409.46.
[0087]
n=3,1hnmr(400mhz,cdcl3):7.96(d,j=8.8hz,2h),7.62(d,j=8.0,hz,1h),7.53-7.48(m,4h),7.42(dd,6h),7.15(dd,4h),5.68-5.59(m,1h),4.92(t,1h),2.85(d,j=8.1hz,2h),1.49-1.88(m,6h).ms[m+h]+=423.49.
[0088]
第七步:参照实施例7方法二的操作,其中n=1,2,3
[0089]
实验编号n反应温度收率1n=125℃至50℃89%2n=225℃至50℃83%3n=325℃至50℃81%
[0090]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1