一种用于医院消毒的苯并噁唑类化合物的制备方法和应用与流程
本发明属于抗菌药物合成技术领域,具体涉及一种用于医院消毒的苯并噁唑类化合物的制备方法和应用。
背景技术:
消毒供应中心是医院内的一个重要科室,是对医院内承担各科室所有重复使用诊疗器械、器具和物品清洗、消毒、灭菌以及无菌物品供应的部门。通过加强复用器械的清洗管理,能够保证医院诊疗活动的安全性,由此可见,消毒供应中心就是医院的“心脏”。对于各类复用器械,必须选用适宜的清洗技术,保证医疗器械质量.
为了减少医院感染几率,必须做好消毒工作。消毒供应中心作为医院的专业消毒部门,在控制医院感染中发挥着重要的作用。当前,随着新技术、新器械的广泛应用,加大了医院感染几率,给医护人员、患者的身心健康带来了危害。同时,很多消毒供应中心依然采用传统的管理模式,不能做到彻底消毒,存在很大的缺陷。
消毒药品是消毒供应中心最常用也是重要的材料,例如现在各大医院常用的季铵盐消毒液,是一类表面活性消毒剂,其消毒杀菌机制是季铵盐阳离子在静电力、氢键力以及表面活性剂分子的作用下聚集到细胞壁上,导致菌体内重要蛋白分子及营养物质外渗,细菌呼吸及糖代谢过程受阻,从而产生室阻效应,导致菌体蛋白变性,抑制细菌生长,导致细菌死亡。铵盐烷基通过与细菌表面蛋白质分子结合,改变细菌胞膜的通透性,继而发生溶胞作用,破坏细菌细胞结构,引起细菌死亡,达到灭菌的效果。
杂环化合物是有机化合物中最庞大的一类,而且在自然界中分布十分广泛,其化学结构也千变万化,各自有着独特的性质和用途。含氮杂环化合物因其具有良好的生物活性而在农药和医药等农业生产和人类健康中发挥着重要的作用,目前已经有许多含氮杂环化合物被开发成农药和医药新产品。苯并噁唑是一类含有n和o杂原子的杂环化合物,该杂环化合物在药物化学中有着广泛的应用,在制药领域中有着广泛的应用前景,该类化合物具有显著的抗肿瘤,抗炎,抗hiv,抗菌以及褪黑素受体拮抗剂等活性。苯并噁唑类衍生物不仅在生物,医药等方面具有广泛的应用,而且在光学材料,高性能复合材料等诸多领域显现出独特的性能。
我们研究团队发明了一种苯并噁唑类的消毒药物,依托医院分子生物实验室和河南湾流生物,发现该类化合物对革兰氏阳性菌和阴性菌均有一定的抑制作用,使其能够溶于乙醇,便于医院消毒中心使用。
技术实现要素:
本发明解决的技术问题是提供了一种操作简单易行、原料廉价易得、反应效率较高且具有良好抑菌效果的苯并噁唑类化合物及其制备方法。
本发明为解决上述技术问题采用如下技术方案,一种用于医院消毒的苯并噁唑类化合物,其特征在于该化合物具有如下结构:
本发明所述的苯并噁唑类化合物的制备方法,其特征在于化合物
(1)、3,4-二氢-1(2h)-萘酮与乙二酸二乙酯在金属钠作用下缩合得到化合物2;
(2)、化合物2与盐酸肼在碱性作用下作用下发生缩合反应得到化合物3;
(3)、化合物3在催化剂作用下与二氯异氰尿酸钠发生反应得到化合物4;
(4)、化合物4与三氟乙醇在碱性环境作用下发生反应得到化合物5;
(5)、化合物5与盐酸肼发生取代反应得到化合物6;
(6)、化合物6和丙酮酸乙酯发生缩合反应得到化合物7;
(7)、化合物7在催化剂作用下发生分子内重排环合后再酯水解得到化合物8;
(8)、化合物8和[2-(1,3-苯并恶唑-2-基)乙基]胺盐酸盐缩合得到目标化合物。
进一步限定,步骤(1)的具体过程为:将一定量的金属钠加入干燥的乙醇中,在氮气保护下加热至回流,可以观察到金属钠逐渐溶解,保持氮气氛围,在50℃条件下,然后通过恒压滴液漏斗缓慢向反应体系中加入一定量的乙二酸二乙酯,滴加完后降至室温,再通过恒压滴液漏斗缓慢滴加溶有3,4-二氢-1(2h)-萘酮的乙醇溶液,反应体系在室温条件下反应一段时间后降至0℃,向反应体系中加入一定二氯甲烷,然后保持0℃条件下缓慢滴加2n的盐酸溶液调节反应体系ph至6~7,浓缩反应液,再向浓缩物中加入二氯甲烷,使其完全溶解,用水洗涤多次后分出有机相,浓缩后经硅胶柱层析分离得到化合物2;所述的3,4-二氢-1(2h)-萘酮与乙二酸二乙酯与金属钠的投料量摩尔比为1:1:2。
进一步限定,步骤(2)的具体过程为:在带有分水器的反应瓶中,将一定量的化合物2加入干燥的溶剂中,再加入一定量的哌啶,碳酸盐和盐酸肼,搅拌均匀后加入沸石,缓慢升温至回流,通过分水器排除反应过程中产生的水分,等分水器中不再有水分出现,继续反应一段时间后趁热过滤反应液,滤液再真空条件先浓缩后加入二氯甲烷,在0℃条件下缓慢滴加2n的盐酸溶液调节反应体系ph至8~9,分出有机相,浓缩后得到化合物3;所述的化合物2与哌啶的投料量质量比为1:1;所述的化合物2与碳酸盐与盐酸肼的投料量摩尔比为1:1:1.5;所述的碳酸盐为碳酸钾或碳酸钠或碳酸铯;所述的反应时间为2-8h。
进一步限定,步骤(3)的具体过程为:在带有氮气保护装置的反应瓶中,把一定量的化合物3加入二氯甲烷和三氟乙酸的混合液中,室温搅拌均匀一段时间后浓缩反应液,然后再加入二氯甲烷和三氟甲磺酸银,在氮气保护下,缓慢滴加溶有二氯异氰尿酸钠的乙腈溶液,滴加完后在氮气保护下缓慢升温至一定温度,保持该温度搅拌反应一段时间后加入一定量磷酸盐,反应结束后降温至0~10℃,在氮气保护下缓慢地滴加水,控制反应体系温度不超过20℃,滴加完后过滤反应液,向反应液中加入活性炭,加热后趁热过滤反应液,然后用稀盐酸调节反应液ph为4~5,加入氯仿搅拌萃取,然后分出有机相;有机相用水洗涤有机相两次,再用饱和氯化钠溶液洗涤一次,最后经无水硫酸镁干燥,抽滤后浓缩,得到化合物4;所述的化合物3与二氯异氰尿酸钠的投料量摩尔比为1:0.6~1;所述的二氯甲烷与三氟乙酸混合液的体积比为v二氯甲烷:v三氟乙酸=2:1;所述的化合物3与三氟甲磺酸银与磷酸盐的投料量摩尔比为10:1:1~1.5;所述的磷酸盐为磷酸钾或磷酸镁;所述的反应温度为60~80℃。
进一步限定,步骤(4)的具体过程为:把一定量的化合物4和一定量的金属醇盐和三氟乙醇加入四氢呋喃中,缓慢升温至回流,tlc监控原料化合物4反应完全,浓缩反应液,再加入二氯甲烷后用水洗涤多次后浓缩,然后在丙酮和正己烷的混合液中中重结晶得到化合物5;所述的金属醇盐为叔丁醇钾或叔丁醇钠;所述的化合物4与金属醇盐与三氟乙醇的投料量摩尔比为1:1~1.5:1.5。
进一步限定,步骤(5)的具体过程为:把一定量的化合物5和三乙胺加入到二甲基亚砜中,在室温条件下加入搅拌均匀,然后缓慢滴加溶有盐酸肼的二甲基亚砜溶液,滴加完后升温至80℃,tlc监控原料反应完全后,降至室温,过滤反应液,向滤液中加入水,搅拌后加入乙酸乙酯萃取多次,合并有机相,在真空条件下浓缩,浓缩物经硅胶柱层析分离得到化合物6;所述的化合物5与三乙胺与盐酸肼的投料量摩尔比为1:1.5:1.1。
进一步限定,步骤(6)的具体过程为:把一定量的化合物6和丙酮酸乙酯加入到甲苯中,在室温条件下,搅拌均匀后加入对甲苯磺酸钠,然后逐渐升温至回流,及时除去反应过程中生成的水,大约回流反应一段时间在氮气保护下向反应液中滴加三氟乙酸,继续回流反应一段时间后停止加热,然后在氮气保护下,向反应液中缓慢加入溶有氢氧化钡的甲醇溶液,滴加完后继续回流搅拌反应一段时间后向滤液中倒入水中,用稀盐酸调节反应液ph为中性,然后用二氯甲烷萃取反应液多次,合并有机相,再经无水硫酸镁干燥后浓缩经硅胶柱层析得到化合物7;所述的化合物6与丙酮酸乙酯与对甲苯磺酸钠的投料量摩尔比为1:1~1.1:1;所述的化合物6与三氟乙酸与氢氧化钡的投料量摩尔比为1:1:2~3。
进一步限定,步骤(7)的具体过程为:把一定量的化合物7和催化剂加入硝基甲烷中,再加入一定量的溴化锂,加热至一定温度,搅拌反应后开起真空,蒸出硝基甲烷,然后加入冰水,搅拌后抽滤反应液,滤饼经丙酮重结晶后加入四氢呋喃中,再加入一定量的氢氧化锂,在加热至回流,反应结束后降至室温,用稀盐酸调节反应液ph为4~5,分出有机相,水相再用二氯甲烷萃取多次,合并有机相浓缩后烘干后得到化合物8;所述的催化剂为bi(otf)3-liclo4;所述的化合物7与催化剂的投料量质量比为5:1~2;所述的反应温度为60~80℃;所述的化合物7与氢氧化锂的投料量摩尔比为1:1~2。
进一步限定,步骤(8)的具体过程为:把一定量的化合物8和[2-(1,3-苯并噁唑-2-基)乙基]胺盐酸盐加入n,n-二甲基甲酰胺中,再加入一定量的hatu和dipea,加热45℃,搅拌反应过夜,tlc监控原料反应完全,真空浓缩,然后加入二氯甲烷,用水洗涤多次,浓缩有机相,浓缩物经正己烷重结晶得到目标化合物;所述的化合物8与[2-(1,3-苯并恶唑-2-基)乙基]胺盐酸盐与hatu与dipea的投料量摩尔比为1:1:1.5:3。
本发明以价格低廉的3,4-二氢-1(2h)-萘酮为起始原料,经过七步常规合成反应,创造性的得到了一种结构新颖的苯并噁唑类化合物,该类化合物对革兰氏阳性菌和阴性菌均具有一定的抑制作用。
附图说明
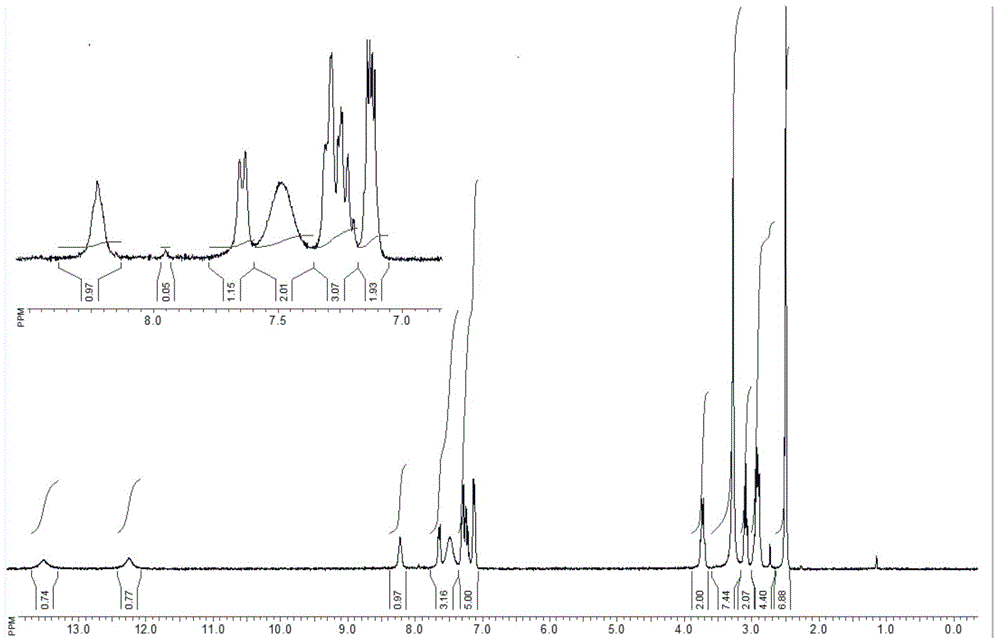
图1目标化合物的核磁氢谱
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
实施例1
将金属钠(4.5g)加入干燥的乙醇200ml中,在氮气保护下加热至回流,可以观察到金属钠逐渐溶解,保持氮气氛围,在50℃条件下,然后通过恒压滴液漏斗缓慢向反应体系中加入乙二酸二乙酯(15g),滴加完后反应一段时间,反应体系降至室温,再通过恒压滴液漏斗缓慢滴加溶有3,4-二氢-1(2h)-萘酮(化合物1,15g)的乙醇溶液200ml,反应体系在室温条件下反应7h,然后降至0℃,向反应体系中加入一定二氯甲烷,然后保持0℃条件下缓慢滴加2n的盐酸溶液调节反应体系ph至6~7,浓缩反应液,再向浓缩物中加入二氯甲烷,使其完全溶解,用水洗涤多次后分出有机相,浓缩后经硅胶柱层析分离得到化合物2(20.8g),lc-ms(esi):m/z247[m+h]+。
实施例2
在带有分水器的反应瓶中,将化合物2(25g)加入干燥的甲苯300ml中,再加入哌啶25g,碳酸钾(14g)和盐酸肼(15g),搅拌均匀后加入沸石2.5g,缓慢升温至回流,通过分水器排除反应过程中产生的水分,等分水器中不再有水分出现,继续反应2h,趁热过滤反应液,滤液再真空条件先浓缩后加入二氯甲烷500ml,在0℃条件下缓慢滴加2n的盐酸溶液调节反应体系ph至8~9,分出有机相,浓缩后得到化合物3(18.6g),lc-ms(esi):m/z243[m+h]+。
实施例3
在带有分水器的反应瓶中,将化合物2(25g)加入干燥的四氢呋喃600ml中,再加入哌啶25g,碳酸铯(32g)和盐酸肼(15g),搅拌均匀后加入沸石2.5g,缓慢升温至回流,通过分水器排除反应过程中产生的水分,等分水器中不再有水分出现,继续反应6h,趁热过滤反应液,滤液再真空条件先浓缩后加入二氯甲烷500ml,在0℃条件下缓慢滴加2n的盐酸溶液调节反应体系ph至8~9,分出有机相,浓缩后得到化合物3(21.1g),lc-ms(esi):m/z243[m+h]+。
实施例4
在带有分水器的反应瓶中,将化合物2(25g)加入干燥的甲苯300ml中,再加入哌啶25g,碳酸钠(10g)和盐酸肼(15g),搅拌均匀后加入沸石2.5g,缓慢升温至回流,通过分水器排除反应过程中产生的水分,等分水器中不再有水分出现,继续反应7.5h,趁热过滤反应液,滤液再真空条件先浓缩后加入二氯甲烷500ml,在0℃条件下缓慢滴加2n的盐酸溶液调节反应体系ph至8~9,分出有机相,浓缩后经硅胶柱层析分离纯化得到化合物3(11.9g),lc-ms(esi):m/z243[m+h]+。
实施例5
在带有氮气保护装置的反应瓶中,把化合物3(24g)加入二氯甲烷400ml和三氟乙酸200ml的混合液中,室温搅拌均匀7h后浓缩反应液,然后再加入二氯甲烷500ml和三氟甲磺酸银(2.6g),在氮气保护下,缓慢滴加溶有二氯异氰尿酸钠(22g)的乙腈溶液250ml,滴加完后在氮气保护下缓慢升温至60℃,保持该温度搅拌反应3h,再加入磷酸钾(2g),可见反应体系逐渐呈现轻微浑浊,tlc监控化合物3反应完全;然后降温至0~10℃,在氮气保护下缓慢地滴加水120ml,控制反应体系温度不超过20℃,滴加完后过滤反应液,向反应液中加入活性炭10g,加热至40℃搅拌20min,趁热过滤反应液,然后用稀盐酸调节反应液ph为4~5,加入氯仿300ml搅拌萃取,然后分出有机相;有机相用水20ml洗涤有机相两次,再用饱和氯化钠溶液80ml洗涤一次,最后经无水硫酸镁干燥,抽滤后浓缩,得到化合物4(15.7g);lc-ms(esi):m/z205[m+h]+;1hnmr(400mhz,dmso-d6):δ12.31(s,1h),7.74-7.71(m,1h),7.29-7.25(m,2h),7.13(d,j=8.0hz,1h),2.92-2.85(m,4h)。
实施例6
在带有氮气保护装置的反应瓶中,把化合物3(24g)加入二氯甲烷400ml和三氟乙酸200ml的混合液中,室温搅拌均匀7h后浓缩反应液,然后再加入二氯甲烷500ml和三氟甲磺酸银(2.6g),在氮气保护下,缓慢滴加溶有二氯异氰尿酸钠(22g)的乙腈溶液250ml,滴加完后在氮气保护下缓慢升温至60℃,保持该温度搅拌反应3h,再加入磷酸镁(4g),可见反应体系逐渐呈现轻微浑浊,tlc监控化合物3反应完全;然后降温至0~10℃,在氮气保护下缓慢地滴加水200ml,控制反应体系温度不超过20℃,滴加完后过滤反应液,向反应液中加入活性炭10g,加热至40℃搅拌20min,趁热过滤反应液,然后用稀盐酸调节反应液ph为4~5,加入氯仿300ml搅拌萃取,然后分出有机相;有机相用水20ml洗涤有机相两次,再用饱和氯化钠溶液80ml洗涤一次,最后经无水硫酸镁干燥,抽滤后浓缩,得到化合物4(17.1g);lc-ms(esi):m/z205[m+h]+;1hnmr(400mhz,dmso-d6):δ12.31(s,1h),7.74-7.71(m,1h),7.29-7.25(m,2h),7.13(d,j=8.0hz,1h),2.92-2.85(m,4h)。
实施例7
在带有氮气保护装置的反应瓶中,把化合物3(24g)加入二氯甲烷400ml和三氟乙酸200ml的混合液中,室温搅拌均匀7h后浓缩反应液,然后再加入二氯甲烷500ml和三氟甲磺酸银(2.6g),在氮气保护下,缓慢滴加溶有二氯异氰尿酸钠(13.5g)的乙腈溶液150ml,滴加完后在氮气保护下缓慢升温至80℃,保持该温度搅拌反应3h,再加入磷酸钾(2g)后继续反应5h;然后降温至0~10℃,在氮气保护下缓慢地滴加水120ml,控制反应体系温度不超过20℃,滴加完后过滤反应液,向反应液中加入活性炭10g,加热至40℃搅拌20min,趁热过滤反应液,然后用稀盐酸调节反应液ph为4~5,加入氯仿300ml搅拌萃取,然后分出有机相;有机相用水20ml洗涤有机相两次,再用饱和氯化钠溶液80ml洗涤一次,最后经无水硫酸镁干燥,抽滤后浓缩,经柱层析分离提纯得到化合物4(9.2g);lc-ms(esi):m/z205[m+h]+;1hnmr(400mhz,dmso-d6):δ12.31(s,1h),7.74-7.71(m,1h),7.29-7.25(m,2h),7.13(d,j=8.0hz,1h),2.92-2.85(m,4h)。
实施例8
在反应瓶中,把化合物4(20g)和叔丁醇钾(11g)和三氟乙醇(15g)加入四氢呋喃300ml中,缓慢升温至回流,反应1h,tlc监控原料化合物4反应完全,浓缩反应液,再加入二氯甲烷300ml,用水50ml洗涤多后浓缩,然后在丙酮和正己烷的混合液中(v丙酮:v正己烷=3:1)300ml中重结晶得到化合物5(24.1g);lc-ms(esi):m/z269[m+h]+;1hnmr(400mhz,cdcl3):δ12.63(s,1h),7.69-7.66(m,1h),7.31-7.28(m,2h),7.17-7.15(m,1h),4.62(s,2h),2.87-2.81(m,4h);元素分析计算值[c13h11f3n2o]:c,58.21;h,4.13;n,10.44.实测值:c,58.12;h,4.16;n,10.38。
实施例9
在反应瓶中,把化合物4(20g)和一定量的叔丁醇钠(14.5g)和三氟乙醇(15g)加入四氢呋喃500ml中,缓慢升温至回流,反应2.5h,tlc监控原料化合物4反应完全,浓缩反应液,再加入二氯甲烷300ml,用水50ml洗涤多后浓缩,然后在丙酮和正己烷的混合液中(v丙酮:v正己烷=3:1)300ml中打浆重结晶洗涤得到化合物5(19.3g);lc-ms(esi):m/z269[m+h]+;1hnmr(400mhz,cdcl3):δ12.63(s,1h),7.69-7.66(m,1h),7.31-7.28(m,2h),7.17-7.15(m,1h),4.62(s,2h),2.87-2.81(m,4h);元素分析计算值[c13h11f3n2o]:c,58.21;h,4.13;n,10.44.实测值:c,58.12;h,4.16;n,10.38。
实施例10
在反应瓶中,把化合物5(26g)和三乙胺(15g)加入到二甲基亚砜300ml中,在室温条件下加入搅拌均匀,然后缓慢滴加溶有盐酸肼(11g)的二甲基亚砜溶液150ml,滴加完后升温至80℃,tlc监控原料反应完全后,降至室温,过滤反应液,向滤液中加入水500ml,搅拌后加入乙酸乙酯100ml萃取5次,合并有机相,在真空条件下浓缩(真空用油泵实现),浓缩物经硅胶柱层析分离得到化合物6(17.4g);lc-ms(esi):m/z201[m+h]+。
实施例11
在反应瓶中,把化合物6(20g)和丙酮酸乙酯(13g)加入到甲苯300ml中,在室温条件下,搅拌均匀后加入对甲苯磺酸钠(2g),然后逐渐升温至回流,及时除去反应过程中生成的水,大约回流反应3h,在氮气保护下向反应液中滴加三氟乙酸(1.2g),继续回流反应1h后停止加热,然后在氮气保护下,向反应液中缓慢加入溶有氢氧化钡(5.0g)的甲醇溶液100ml,滴加完后继续回流搅拌反应3h,然后向滤液中倒入水200ml中,用稀盐酸调节反应液ph为中性,然后用二氯甲烷50ml萃取反应液多次,合并有机相,再经无水硫酸镁干燥后浓缩经硅胶柱层析得到化合物7(24.8g);lc-ms(esi):m/z299[m+h]+。
实施例12
在反应瓶中,把化合物7(30g)和bi(otf)3-liclo46g加入硝基甲烷600ml中,再加入溶有溴化锂0.9g的硝基甲烷溶液50ml,然后加热至75℃,搅拌反应1h后趁热过滤反应液,然后开起真空蒸除硝基甲烷,然后加入冰水1000ml,搅拌10min后抽滤反应液,滤饼经丙酮350ml重结晶洗涤后加入四氢呋喃500ml中,再加入氢氧化锂4.8g,在加热至回流,反应3h后降至室温,用稀盐酸调节反应液ph为4~5,分出有机相,水相再用二氯甲烷50ml萃取多次,合并有机相浓缩后烘干后得到化合物8(17.4g);1hnmr(400mhz,cdcl3):δ12.79(s,1h),8.36(s,1h),7.69(d,j=12.0hz,1h),7.25-7.19(m,2h),7.11(d,j=4.0hz,1h),2.94-2.87(m,4h)。
实施例13
在反应瓶中,把化合物7(30g)和bi(otf)3-liclo412g加入硝基甲烷800ml中,再加入溶有溴化锂0.9g的硝基甲烷溶液50ml,然后加热至80℃,搅拌反应1h后趁热过滤反应液,然后开起真空蒸除硝基甲烷,然后加入冰水1000ml,搅拌10min后抽滤反应液,滤饼经丙酮400ml重结晶洗涤后加入四氢呋喃500ml中,再加入氢氧化锂4.8g,在加热至回流,反应3h后降至室温,用稀盐酸调节反应液ph为4~5,分出有机相,水相再用二氯甲烷50ml萃取多次,合并有机相浓缩后烘干后得到化合物8(22.7g);1hnmr(400mhz,cdcl3):δ12.79(s,1h),8.36(s,1h),7.69(d,j=12.0hz,1h),7.25-7.19(m,2h),7.11(d,j=4.0hz,1h),2.94-2.87(m,4h)。
实施例14
在反应瓶中,把化合物8(25g)和[2-(1,3-苯并噁唑-2-基)乙基]胺盐酸盐(20g)加入n,n-二甲基甲酰胺1200ml中,再加入hatu(57g)和dipea(40g),加热45℃,搅拌反应过夜,tlc监控原料反应完全,真空浓缩,然后加入二氯甲烷1000ml,用水200ml洗涤多次,分出有机相后浓缩,浓缩物经正己烷重结晶得到目标化合物34g;lc-ms(esi):m/z398[m+h]+;1hnmr(400mhz,dmso-d6):δ13.51(s,1h),12.22(s,1h),8.43(s,1h),7.64(d,j=8.0hz,1h),7.47(s,2h),7.29-7.16(m,3h),7.13-7.10(m,2h),3.69(d,j=4.0hz,2h),3.08(t,j1=8.0hz,j2=8.0hz,2h),2.89-2.78(m,4h)。
实施例15
抗菌活性测试:通过牛津杯琼脂扩散法测试了苯并噁唑类化合物对大肠杆菌、金黄葡萄球菌的抑菌活性;配制苯并噁唑类化合物浓度为1mg/ml的二甲基亚砜溶液,以浓度为1mg/ml的比西林的二甲基亚砜溶液为阳性对照,溶剂二甲基亚砜为空白对照;各样品均重复5次与37℃培养24h,在培养中,一方面试验菌开始生长,另一方面抗生素呈球面扩散,离杯越近,抗生素浓度越大,离杯越远抗生素浓度越小。随着抗生素浓度减小,有一条最低抑菌浓度带,在带范围内细菌不能生长,而呈透明的圆圈,这就叫“抑菌圈”,抑菌直径取其平均值。
经重复五次实验,求平均值后所得到的苯并噁唑类化合物对大肠杆菌的抑菌圈直径为22.97mm,对金黄色葡萄球菌的抑菌圈直径为24.72mm,均优于比西林(14.7mm和22.5mm)。
以上实施例仅为说明本发明的技术思想,不能以此限定本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案基础上所做的任何改动,均落入本发明保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!