在确定性限制性位点全基因组扩增(DRS-WGA)之后分析杂合性缺失(LoH)的方法与流程
在确定性限制性位点全基因组扩增(drs-wga)之后分析杂合性缺失(loh)的方法
1.相关申请的交叉引用
2.本专利申请要求于2019年7月30日提交的意大利专利申请第102019000013335号的优先权,其全部公开内容通过引用并入本文。
技术领域
3.本发明涉及在使用或不使用正常对照的情况下由来自确定性限制性位点全基因组扩增(drs-wga)的低通全基因组测序数据分析样本中的杂合性缺失(loh)的方法,从而实现单细胞分辨率。该方法可应用于数种单细胞应用,例如肿瘤学,包括分析循环肿瘤细胞以及组织样本中的单细胞异质性;或生殖医学,包括植入前基因筛查(pgs)。
背景技术:
4.通常需要对单细胞基因组dna进行全基因组扩增(wga)来获得更多dna,以简化和/或允许包括测序、snp检测等在内的不同类型的遗传分析。基于确定性限制性位点使用lm-pcr进行的wga(下文称为drs-wga中)从wo2000/017390中获知。
5.重要的是,drs-wga在许多方面都被表明是同类最佳的wga方法,特别是在单细胞的较低等位基因漏失方面(borgstrom等人,2017;normand等人,2016;babayan等人,2016;binder等人,2014)。
6.hodgkinson c.l.等人,自然医学(nature medicine)20,897
–
903(2014)中使用了基于lm-pcr的drs-wga商业试剂盒(ampli1
tm wga试剂盒,silicon biosystems)。在这项工作中,通过对单细wga材料进行低通全基因组测序的拷贝数分析,进行wga衔接子的消化和片段化,然后进行有条形码的illumina接头连接以测序。
7.wo2017/178655和wo2019/016401a1教导了从drs-wga(例如ampli1)或malbac制备大规模平行测序文库用于低通全基因组测序和拷贝数分析的简化方法。在ferrarini等人,plosone 13(3):e0193689https://doi.org/10.1371/journal.pone.0193689中,已参考拷贝数分析对使用ion torrent平台的wo2017/178655的方法的性能进行了详细说明.
8.ampli1
tm wga与阵列比较基因组杂交(acgh)兼容。确实有几组(moehlendick b等人,2013,plos one 8(6):e67031;czyz zt,et al.,2014,plos one 9(1):e85907)表明它适用于高分辨率拷贝数分析。然而,acgh技术昂贵且劳动强度大,使得可能需要不同的方法,例如用于检测体细胞拷贝数改变(cna)的低通全基因组测序(lpwgs)。
9.当使用阵列cgh、中期cgh以及其他遗传分析测定(例如使用靶向引物的杂合性缺失和用于分析选定的微卫星的pcr)时,已显示drs-wga比dop-pcr更好地分析来自微量显微切割ffpe材料的拷贝数谱(stoecklein等人,am j pathol.2002jul;161(1):43-51;arneson等人,isrn oncol.2012;2012:710692.doi:10.5402/2012/710692.epub 2012mar 14.)。
10.us 7,424,368 b2教导了一种估计实验样本中基因组区域的拷贝数的方法,该方
90%)下loh评分的接受者操作特征(roc)曲线下面积(auc)值。
43.图14示出了从使用illumina的ampli1 lowpass制备的测序文库中获得的与片段大小选择相关的数据。
44.图15示出了通过使用恒定碱基对基因组窗口和应用fisher统计检验来检测loh的示例。
45.图16示出了通过使用具有恒定数量基因座的基因组窗口来检测loh的示例。
46.图17示出了用于识别loh评分阈值的基于9个单细胞的训练集创建的roc曲线,loh评分阈值被定义为n=1000个snp的窗口中的杂合基因座数量,这1000个snp被至少1个读段与1000000个映射读段所覆盖。
47.图18示出了根据本发明实施例2中的方法检测的单个肿瘤细胞和loh基因组区域的图。
48.图19示出了使用拷贝数分段和fisher检验来检测具有loh状态的基因组区域的示例。
49.定义
50.除非另有定义,否则本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解相同的含义。尽管在本发明的实践或测试中可以使用与本文所述的那些相似或等效的许多方法和材料,但在下文中描述了优选的方法和材料。除非另有说明,本文描述的用于本发明的技术是本领域普通技术人员熟知的标准方法。
51.表述“大规模平行下一代测序(ngs或mps)”是指一种对dna进行测序的方法,该方法包括创建在空间和/或时间上分离、克隆测序(先前进行或未进行克隆扩增)的dna分子文库。实例包括illumina平台(illumina inc)、ion torrent平台(thermo fisher scientific inc)、pacific biosciences平台、minion(oxford nanopore technologies ltd)。
52.表述“低通全基因组测序”是指参照整个参考基因组以低于3的平均测序深度进行全基因组测序。
53.表述“平均测序深度”在此意指在每个样本的基础上,测序并映射到参考基因组的碱基总数除以总参考基因组大小。测序和映射的碱基总数可以近似为映射读段数量乘以平均读段长度(the number of mapped reads time the average read length)。
54.表述“参考基因组”是指特定物种的参考dna序列。
55.术语“基因座(locus)”(复数“基因座(loci)”)是指染色体上的固定位置(相对于参考基因组)。
56.表述“多态基因座”是指具有2个或更多个等位基因的基因座,在群体中观察到的频率大于1%。
57.表述“杂合基因座”是指在特定样本中观察到的具有2个或更多个等位基因的基因座。
58.表述“基因组窗口”是指包含在单个染色体中的参考基因组的区间,其具有固定或可变长度。
59.表述“基因组区域”是指包含同一染色体中的一个或多个相邻基因组窗口的区间。
60.表述“覆盖的基因组”意指被至少一个读段覆盖的参考基因组的部分。
61.术语“读段(read)”是指由测序仪测序(“读取(read)”)的dna片段(piece of dna)。
62.表述“拷贝数区域”意指与相同拷贝数值相关的基因组区域。
63.表述“分段拷贝数区域”意指作为cna的生物信息学分析结果,与相同拷贝数值相关的基因组区域。
64.表述“肿瘤抑制基因”是指由于例如序列变体(种系或体细胞)导致其功能丧失而与肿瘤发生概率增加有关的基因。
65.表述“减少率”是指根据drs-wga中使用的限制性酶对参考基因组进行计算机消化获得的包括在特定碱基对范围内片段的碱基总数除以参考基因组中的碱基总数。
66.表述“杂合性缺失”或“loh”意指基因组区域中等位基因之一缺失。
67.表述“loh调用(loh call)”是指对loh(在基因组区域中)存在的指派。
68.表述“等位基因含量”意指在基因座处检测到的等位基因方面的组成。
69.除非另有说明,为简单起见,在本发明的描述中,如果仅检测到一个等位基因,则基因座将可互换地称为纯合或单等位基因,而在存在至少两个等位基因的情况下,基因座将被称为杂合或双等位基因,无论该基因座的真实基因型如何。
具体实施方式
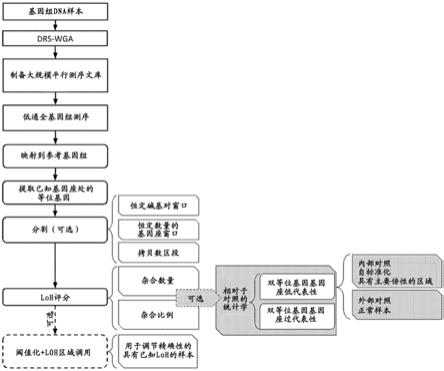
70.参考图1,根据本发明的分析包含基因组dna的至少一个样本中的杂合性缺失(loh)的方法包括以下步骤。
71.在步骤a中,提供包含基因组dna的至少一个样本。
72.在步骤b中,进行所述基因组dna的确定性限制性位点全基因组扩增(drs-wga)。
73.在步骤c中,由所述drs-wga的产物制备大规模平行测序文库。
74.在步骤d中,对所述大规模平行测序文库以《1、优选《0.05、更优选《0.01的平均覆盖深度进行低通全基因组测序。
75.在步骤e中,将步骤d中获得的读段在所述至少一个样本的参考基因组上进行比对。
76.在步骤f中,提取多个基因座处的等位基因含量。所述多个基因座包括多态基因座和/或杂合基因座。
77.在步骤g中,根据所述多个基因座中具有至少两个不同等位基因的基因座的数量将loh评分指派给针对所述至少一个样本的所述参考基因组的至少一个基因组窗口。
78.优选地,在制备大规模平行测序文库的步骤c之前、过程中或之后进行大小选择步骤,并且制备大规模平行测序文库的步骤不包括随机片段化步骤。
79.大小选择步骤优选保留100至800个碱基对范围内的片段。
80.在本发明的某些实施方式中,大小选择优选保留300至450个碱基对范围内的片段。
81.在本发明的某些实施方式中,在大小选择步骤中保留的片段的峰值优选集中在150bp至600bp的碱基对范围,更优选地,大小选择步骤保留425至575个碱基对范围内的片段。
82.优选地,至少一个基因组窗口:
[0083]-具有恒定的碱基对宽度,或
[0084]-具有恒定数量的所述多个基因座,或
[0085]-选自由染色体、染色体臂和分段的拷贝数区域组成的组。
[0086]
多个基因座优选地包含从针对所述至少一个样本的参考基因组或通过对一组参考样本进行基因分型而获得的数据库(例如dbsnp)中获得的多态基因座。
[0087]
作为替代方案,多个基因座优选地包含用于对照样本的已知杂合基因座。
[0088]
当基因组窗口具有恒定的碱基对宽度或具有恒定数量的多个基因座,或者多个基因座包含针对所述样本的参考基因组的多态基因座时,loh评分优选对应于在所述至少一个基因组窗口中的杂合基因座的数量。
[0089]
优选地,loh评分对应于至少一个基因组窗口中杂合基因座占多态基因座总数的比例。
[0090]
loh评分优选地对应于统计检验的p值。
[0091]
统计检验优选地评估双等位基因座相对于测序和wga错误率的过代表性(over-representation)的显著性或双等位基因座相对于对照样本的低代表性(under-representation)的显著性。
[0092]
对照样本优选包含来自至少一个样本的主要倍性的至少一个基因组区域。
[0093]
对照样本优选是至少一个正常样本,该样本更优选从获得所述至少一个样本的同一待测个体获得。在肿瘤学的情况下,对照样本优选是正常(非肿瘤)样本。
[0094]
在循环胎儿细胞的情况下,对照样本优选为母本样本。可替代地,当父本样本可用时,它可以是父本样本或母本样本和父本样本的组合。可以利用母本和/或父本基因型的可用性来选择已知在所述亲本对照中为杂合的基因座子集。
[0095]
优选地,如果所述loh评分超过(pass)基因组窗口的阈值,则将所述基因组窗口称为处于loh中。在这种情况下,该方法更优选地包括:如果包含在至少一个基因组区域中的每个基因组窗口的loh评分超过所述阈值,则将loh状态指派给该区域的步骤;或者根据包含在至少一个基因组区域中的基因组窗口的loh状态将loh状态指派给该区域的步骤。
[0096]
更优选地,所述至少一个基因组区域包含肿瘤抑制基因,其甚至更优选选自由brca1、brca2、palb2、tp53、cdkn2a、rb1、apc、pten、cdkn1b、dmp1、nf1、aml1、egr1、tgfbr1、tgfbr2和smad4组成的组。
[0097]
至少一个样本优选具有至少50%的纯度。更优选地,所述至少一个样本是单细胞。
[0098]
drs-wga中片段长度单义关系的基因座
[0099]
更详细地,根据本发明的方法利用了这样一个事实,即在drs-wga,例如ampli tm wga中,基因组中的每个基因座在wga文库中仅以具有特定碱基对长度的片段表示。该特性可以被指定为“基因座到片段长度的单义关系”(l2flur)。考虑一般的正常基因座,例如多态性snp的基因座,所述基因座将仅以给定长度的片段表示,给定长度等于限制酶消化后相应片段的大小(在任一单链上测量),加上通用wga衔接子长度(在ampli1 wga的情况下即lib1引物的长度)的两倍。当根据ampli1 lowpass试剂盒制备文库后对wga进行测序时,会引入与已知的测序衔接子和条形码长度相关的可预测的额外长度。
[0100]
诸如未消化的限制性位点或序列变体的非理想因素以及其他因素可能会相对于理论上的预期影响和扭曲wga产物中给定片段的代表性频率。这些因素通常是适度的,而且
只要它们是可重复的,则它们的非随机性质可以通过补偿它们的影响来部分抵消。因此,除非另有说明,否则在本说明书中将忽略它们的影响。
[0101]
基因组的代表性减少
[0102]
在根据本发明的方法中,利用l2flurl特性来产生基因组的代表性减少,由此,对于给定的读段数量,低通测序数据通过相对于样本参考基因组的原始大小有效减小覆盖基因组的大小,实现了对覆盖基因组的有效的更高的覆盖。换句话说,wga片段的大小选择产生了参考基因组的确定性下采样(deterministic subsampling)。术语“确定性”是必不可少的,因为-增加读段数量-最终重新采样相同的基因组位点(见图2)。
[0103]
图2示出了基因组代表性减少对观察到的覆盖率的影响。图2a示出了根据3种不同方法的msei片段长度分布:进行大小选择收集300至450bp的片段的用于ion torrent的ampli1 lowpass(a1lp_ss)、进行测序步骤产生的选择的ampli1 lowpass(a1lp)以及ampli1 wga后进行随机片段化和测序之后获得的文库(a1_wfrg)(binder v等人,2014)。这3种不同的方法代表了基因组代表性的降低的不同水平,从最严格的a1lp_ss到以缺乏选择为特征的a1_wfrg。图2b显示了使用不同方法获得的lorenz曲线,显示覆盖均匀性随着大小选择水平逐渐降低。较低的a1lp_ss均匀性可以通过dna模板的饱和度和相同片段的重复测序来解释。模板饱和度由图2c和2d中的图确认,它们分别以映射读段的增量间隔显示覆盖碱基的总量和每个碱基的平均覆盖率。这些图清楚地表明,大小选择步骤(a1lp_ss)减少了可用dna的数量,其效果是有限的覆盖靶标,但覆盖率更高。
[0104]
值得注意的是,该方法是灵活的,因为取决于所需的分辨率和/或测序平台和使用的测序方案,不同的确定性酶可能是合适的。例如,可以使用不同的高频剪切酶(frequent cutter)。在ampli1 wga的示例中,ttaa基序是限制性位点。其他四碱基剪切酶可用于在不同的限制性位点切割,例如gtac、ctag(图3),从而获得不同的片段分布。图3示出了具有不同限制性位点(四个或六个碱基对)的人类基因组的计算机消化。对于给定的片段长度范围(例如,适用于某种测序仪和大小选择方法),不同的限制性位点会产生不同数量的片段。
[0105]
在初次pcr后首次纯化drs-wga时,会进行第一次大小选择,从而将wga的较短片段与游离引物一起去除。有利地,该方法使用进一步的选择步骤。这个额外的选择步骤可以通过从初次wga中选择某些片段的大小和/或通过限制可测序片段的方法生成大规模平行测序文库来实现。例如,ampli1lowpass试剂盒包含固有的大小选择步骤,足以对该方法产生积极影响。在wo2017/178655中,对凝胶进行大小选择。在wo2019/016401中,使用spri珠子的连续纯化步骤有效地产生了第一尺寸选择,由此碱基对的长度被限制在基本上取决于spri珠子浓度的范围内。此外,测序仪本身也可以引入大小选择,因为较长的片段会以越来越低的效率生成序列数据(例如,由于ion torrent中的乳液pcr效率,或illumina平台中用于簇形成的桥式pcr)。
[0106]
在drs-wga中,测序文库的平均大小与参考基因组的下采样率之间也存在确定性关系。
[0107]
对人类参考基因组hg19的ttaa消化进行的计算机分析(图4)产生了包括所有染色体序列在内的总共约19m片段,这将转化为正常二倍体人类基因组上的38m片段。例如,计算机选择175至225bp范围内的片段将只有1252559个,其覆盖3.09b个碱基中的共计大约248m个碱基,即人类参考基因组的8.02%。参见下表1,其中按大小选择的不同范围列出了片段
数、总碱基对和缩减率(%)。这种下采样可以指定为缩减率(rr)。
[0108]
表1
[0109]
片段之间的间距
[0110][0111]
除了缩减率之外,在drs-wga中,连续片段之间的平均间距也存在确定的关系,这取决于选择用于测序的片段长度分布的部分。在这方面,参见图5,其中图a示出了对具有
±
100bp的带(band)的三个不同的片段大小200、500、800进行测量,由于选择的片段数量减少,片段长度和间距之间正相关;b图示出了,对于每个片段大小,使用三个不同的带(
±
50、
±
100、
±
150)证明仍然由于可获得的具有更窄大小范围的片段数量减少,带大小和间距之间负相关。
[0112]
一般地,通过人类参考基因组hg19关于ampli1 drs-wga片段分布的计算机分析,发现:
[0113]
●
所选片段的平均碱基对长度越高,片段数量越少,它们之间的间距越大;
[0114]
●
选择的片段范围越窄,片段的数量就越少,它们之间的间距越大。
[0115]
片段大小选择
[0116]
也可以使用不同的大小选择技术来实现所需的缩减率,这取决于每个样本和/或分辨率中所选择的测序读段数量。参考图4,明显的是,对于给定的平均片段长度,选择集中于该平均片段长度的分别更小或更大的带可以获得更小或更大数量的总片段。
[0117]
像pipping prep(sage science)这样的仪器可用于对片段长度分布进行更严格的控制,并且使用通带滤波器类似物,还可以具有更高的q因子,q因子被定义为
[0118]
q=fcenter/deltaf=[(fmin+fmax)/2]/(fmax-fmin)
[0119]
其中
[0120]
fcenter=(fmin+fmax)/2是片段的平均大小
[0121]
deltaf=fmax-fmin是片段大小范围的宽度
[0122]
fmin是片段大小,低于该大小的片段以常规相对水平(例如1/10=10%)表示或相对于每个容器(bin)中的标准化的带内峰值片段数更小。
[0123]
fmax是片段大小,高于该大小的片段以常规相对水平表示或相对于每个容器(bin)的标准化的带内峰值片段数更小。
[0124]
对于illumina测序,测序模式优选是配对末端测序,因为覆盖的基因组增加,因此每百万读段对的基因座数量增加,从而提高了分辨率。然而,当选择用于测序的大小低于某个大小时,由于两个配对读段完全重叠,因此配对末端测序不会增加覆盖率。
[0125]
使用ion torrent测序,更高的读段长度将成比例地增加覆盖的基因组,因此每百万读段的基因座数量增加,从而提高分辨率。在ampli1 lowpass iontorrent试剂盒(menarini silicon biosystems)中,在凝胶上或使用pippin prep等其他方法对带条形码的池化样本进行大小选择。选择不同的q因子和平均片段长度可以在每百万个读段的基础上提供不同的分辨率。
[0126]
池化样本并选择文库大小进行随后测序的一个优点是所有样本将具有相同的片段长度分布,反过来这将使不同样本中覆盖的基因组的重叠最大化。这在使用基于对照(例如正常对照或母本对照)的方法来识别待测样本(sut)中的潜在杂合基因座时是相关的。
[0127]
另一方面,当使用用于illumina的ampli1 lowpass试剂盒时,首先对不同的lowpass文库进行大小选择,然后将其池化,从而在不同样本中获得略微不同的大小选择,从而在每百万个读段的基础上减少不同样本中覆盖的基因组。尽管标准方案没有强制要求,但文库池化后的大小选择可用于增加样本之间的重叠,这可能有利于基于对照的分析。
[0128]
根据本发明,drs-wga和lpwgs的组合出人意料地导致输入样本的代表性减低。通过使用ngs进行测序,这种参考基因组的降低的代表性文库反过来又缩小了所选(或任何可测序的)碱基对范围内的覆盖的基因组,并且与使用随机启动或随机剪切的替代wga方法相比,在获得了每百万个读段的基础上对覆盖的基因组的有效的更高覆盖。
[0129]
根据本发明,可以根据情况以不同的方式利用这种效果。
[0130]
示例是一个或多个对照样本的可用性——例如“匹配的正常”和一个或多个待测样本(sut)(例如肿瘤样本)的可用性。在这种情况下,drs-wga增加了sut和对照之间的读段重叠。
[0131]
另一个例子是无对照情况,如植入前基因筛查(pgs)的情况,在这种情况下只有与sut对应的单个样本可用。在这种情况下,drs-wga会增加由多于一个读段所覆盖的基因座数量。
[0132]
优选地,从drs-wga制备文库是wo2017/178655和wo2019/016401中公开的方法之一,因为与如在binder v等人,2014或hodgkinson cl等人,2014中进行的消化wga接头、使dna片段化和随后创建可测序文库相比,所得缩减率更高。事实上,dna剪切增加了原始drs-wga的可能的不同片段的数量,这些片段可见于选择用于测序的给定碱基对范围内,因为一旦片段化,较长的片段将回落到上述范围内,而由于片段化,只有一小部分原本在范围内的初级wga片段将被剔除范围,因为相对于较长的片段,较小的片段往往剪切效率较低(参见图2)。
[0133]
loh分析
[0134]
再次参考图1,大规模平行测序文库优选使用ampli1 lowpass试剂盒(用于ion torrent或用于illumina)获得。使用兼容的测序仪对样本进行测序。将从所述文库获得的测序读数映射到参考人类基因组,并提取已知基因座和/或多态基因座中存在的等位基因。优选地,此类基因座被至少2个测序读段覆盖。应注意的是,单个等位基因的检测并不一定意味着真正的纯合基因型,而可能是测序覆盖率低的结果。所述多个基因座优选地根据基因组分区的不同标准在基因组窗口中细分。这种分区是任选的,因为在某些实施方式中,人们可能只对一个或几个预定基因组窗口的分析感兴趣,例如单个染色体或包含一个或多个感兴趣基因的单个基因组基因座。在基因组窗口中检测到的基因座的等位基因状态用于获
得测量值。这种测量值,以下称为loh评分,可以通过根据本发明的多种方法获得,例如对基因组窗口中杂合基因座的数量进行计数,或计算杂合基因座的比例。此外,通过与内部对照比较或通过使用外部对照(来自相同个体或来自不同个体),优选应用统计检验来确定与loh事件相对应的杂合基因座下降(drop)的显著性。可替代地,优选地应用统计检验来确定相对于基于测序和wga错误率的预期,对应于不在loh中的基因组区域的杂合基因座过代表性的显著性。最后,基于从具有已知loh事件的训练数据集计算的固定阈值,优选应用loh评分阈值化来定义对应于loh事件的基因组区域。下面详细介绍该方法的单个步骤。
[0135]
基因组分区(genomepartitioning)
[0136]
参考图1,分区的可选步骤可以三种可替代的方式执行:
[0137]
i)恒定碱基对基因组窗口
[0138]
ii)基因座窗口的恒定数量
[0139]
iii)拷贝数区段
[0140]
在图6所示的替代方案i)中,基因组窗口具有恒定宽度。每个基因组窗口包含多个基因座,其数量取决于基因组位置。当将样本与一组对照正常样本进行比较时,这种方法可能是有利的,因为参考基因组在所有样本中以相同的方式分区,从而允许跨多个样本直接比较每个基因组窗口的loh评分。由于在定义宽度的基因组窗口中检测到的杂合基因座的数量和比例将在更高的读段深度下增加,以允许将样本与对照(或多个)对照样本进行比较,每个样本中映射读段数量优选被标准化为固定的读段数量。这种标准化是通过随机采样读段,映射到参考基因组,直到达到所需的次数来执行的。标准化的读段数量可以是例如100万或200万个读段,优选300万、400万、500万、600万、700万、800万或900万个读段。
[0141]
图6示出了基于恒定碱基对基因组窗口分区的示例的示意图。展示了成对的对照(顶部)和测试(底部)样本。实线代表(一部分)基因组。菱形标记界定了恒定宽度的基因组窗口,已知的多态基因座由点表示(杂合基因座:白色填充点;纯合基因座:灰色填充点)。每个基因组窗口检测到的基因座数量在整个基因组中有所不同,但平均而言,对于给定窗口,预计在两个不同样本之间是相似的,其总读段映射已被标准化为定义的读段计数。与正常对照样本中的相同窗口相比,测试样本中loh中的基因组窗口预计会显示杂合基因座的下降。由于沿基因组的snp密度存在偏差,同一窗口不能直接与位于同一(或其他)样本上不同基因组位置的基因组窗口进行比较。
[0142]
在图7所示的替代方案ii)中,基因组窗口具有恒定数量的基因座。这种方法允许跨整个基因组针对不同snp密度的loh评分进行标准化。当使用无对照方法时,该方法可能是有利的,因为它允许例如对所有基因组窗口应用相同的阈值,而不管它们在基因组中的位置和它们潜在的snp密度。当将测试样本与对照样本进行比较时,该方法可能是不利的,因为根据采样和低通测序检测的基因座分布,对不同的样本可能生成不同的基因组窗口。
[0143]
图7示出了基于每个窗口基因座的恒定数量分区的示例的示意图。展示了成对的对照(顶部)和测试(底部)样本。实线代表(一部分)基因组。菱形标记界定了包含恒定数量基因座的基因组窗口。已知的多态基因座由点表示(杂合基因座:白色填充点;纯合基因座:灰色填充点)。由于测序覆盖率低,不会检测到基因组区域中的所有基因座。因此,基因组窗口末端可以基于通过测序读段进行的基因座采样在不同样本之间变化,因此,在测试样本中检测到的基因组窗口不能直接与其他(对照)样本中的相应基因组窗口进行比较。相对于
不在loh中的同一样本的基因组窗口,预期测试样本中loh中的基因组窗口显示杂合基因座的下降。
[0144]
在具有恒定数量基因座的基因组窗口中检测到的杂合基因座的数量和比例会随着读段深度的增加而增加(参见图8)。优选地,为了允许将loh评分阈值化为预先计算的值,每个样本中映射的读段数量被标准化为固定的读段数量。这种标准化是通过随机采样读段,映射到参考基因组,直到达到所需的次数来执行的。标准化的读段数量可以是例如100万或200万个读段,优选300万、400万、500万、600万、700万、800万或900万个读段。
[0145]
在如图9所示的替代方案iii)中,基因组窗口是两个拷贝数断点之间的分段基因组区域,其被包含在染色体臂中,可以通过根据gc含量标准化基因组窗口中的原始拷贝数计数(boeva,v.等人,2011,bioinformatics,27(2),268
–
269)和通过应用分割算法例如基于lasso的算法(harchaoui,z.等人,2008,adv.neural inform.process.syst.,20,617
–
624)、循环二进制分割(cbs)(seshan ve.等人,2019,dnacopy:dnacopy number data analysis.r package version 1.58.0)或类似的标准化读段计数的算法来定义。该方法基于这样的假设,即相对于样本主要“正常”倍性而言,显示拷贝数水平变化的基因组区域可能受到单个基因组拷贝数畸变事件的影响,因此预期具有统一的loh状态。与替代方案(i)和(ii)相比,由该方法定义的基因组窗口通常要大得多(高达2-3个数量级),并且将包含大量已知的杂合和/或多态基因座,因此能够获得更高的统计功效。此外,通过结合2个不同的生物学维度(拷贝数、loh评分),使用该方法可以获得更准确的结果和更低的假阳性率。然而,在用该方法仍然无法检测到的较小的loh事件位于较大的拷贝数事件中的情况下,该方法可能是不利的。由于染色体臂在复制后经历loh事件并不少见,因此优选地,染色体臂将用作染色体中的分割单元,而不改变拷贝数。这防止了当只有较长的臂受到影响时将较短的染色体臂误作为loh调用(假阳性),或者在双重情况下,当只有较短的染色体臂受到影响时,将较短的染色体臂误作为没有-loh调用(假阴性)。
[0146]
更具体地,图9提供了受两个拷贝数改变事件影响的染色体臂(基因组主要倍性=2)的拷贝数谱的示例性代表:拷贝数丢失段,拷贝数=1;拷贝数增益,拷贝数=3。基因组窗口定义为2个连续拷贝数断点之间的区域。
[0147]
也可以采用分割,利用拷贝数信息来排除源自高水平扩增的假阳性。事实上,高水平扩增最可能源自单个等位基因,因此在该区域的等位基因代表性中引入了偏差,从而次要等位基因(即便存在)将被低代表性并可能导致假阳性loh调用。
[0148]
下表2示出了根据本发明的每个可替代的分区步骤的主要特征和优缺点。
[0149]
表2
[0150][0151]
loh评分
[0152]
根据在所述多个基因座中具有至少两个不同等位基因的基因座的数量将loh评分指派给针对所述至少一个样本的所述参考基因组的至少一个基因组窗口的步骤g也涉及替代的优选实施方式。
[0153]
在一种优选实施方式中,loh评分对应于所述至少一个基因组窗口中杂合基因座的数量。与不在loh中的区域或样本相比,loh中的基因组窗口预计会显示杂合基因座稀缺(参见图10)。
[0154]
在另一种优选实施方式中,对于每个基因组窗口,loh评分被定义为在该基因组窗口中检测到的杂合基因座相对于同一基因组窗口中的多态基因座总数的比例(图11)。与上述方法类似,在存在loh事件的情况下,预期loh评分会持续降低。当窗口不包含检测到的同质数量的基因座时,例如当使用恒定碱基对基因组窗口或使用拷贝数区段来对基因组分区时,该方法可能是有利的。
[0155]
loh评分-统计检验
[0156]
优选地,对于每个基因组窗口,loh评分由对观察到的双等位基因座频率的统计检验的结果定义。
[0157]
在优选实施方式中,杂合基因座的低代表性相对于内部/外部对照的显著性可以通过进行统计检验来评估。具体来说,考虑以下两种分类,为每个基因组窗口建立一个列联表:1)样本类型(测试、对照);2)基因座类型(杂合、纯合)。然后应用统计检验,例如用于分析列联表的fisher精确检验或比对检验(例如:卡方检验、g检验、barnard精确检验、
fisher-freeman-halton检验)。优选地,统计检验应该单侧进行,以便将检测限制在由于loh导致杂合基因座低代表性的情况。事实上,当在给定的基因组区段中存在增益时,即拷贝数增加时,使用低通wgs的读段数量会增加。这可能会导致在没有loh的情况下出现更多的杂合基因座,并且可能会通过双侧统计检验但出于与分析目的相反的原因而标记为显著。
[0158]
在可替代的优选实施方式中,可以测试杂合基因座的过代表性相对于测序和wga错误率的预期的显著性。这种方法在测试单倍体单细胞(例如配子)中的“杂合性增益”(以下简称goh)时可能是有利的。这可能例如由于在减数分裂期间导致染色体增益的不平衡分离中的错误而发生。
[0159]
鉴于针对每个实验执行的大量测试(对于具有500、1000和1500个snp的固定窗口的100万读段样本,大约200、400、600次),可以应用多重测试校正(参见例如benjamini y等人,1995,皇家统计学会杂志(journal of the royal statistical society).系列b(方法论)vol.57,no.1:pp.289-300)。然后将loh评分定义为统计检验得出的p值。
[0160]
对照样本
[0161]
对照可以是“内部的”并且可以例如通过考虑其倍性等于最可能的主要(平均)基因组倍性的基因组区域来定义。这种方法假设大多数不显示拷贝数改变的基因组区域不在loh中。
[0162]
可替代地,对照可以是“外部的”并且可以例如通过使用来自同一待测个体或来自不同个体的一个或多个正常样本来产生。
[0163]
内部对照的使用对于二倍体或多倍体样本(例如:肿瘤样本)可能是有利的,因为它不依赖于读段数量(不需要对映射读段数量进行标准化),并且在样本受损的情况下(例如:ffpe样本)可能是有利的。事实上,与未受损的样本相比,受损样本可能显示出更高的丢弃(dropouts)的发生,其中基因座的两个等位基因之一由于dna损伤而丢失,因此,对于不在loh中的基因组区域,杂合位点的数量低于预期。这可能会阻碍测试与具有不同破坏程度的外部对照样本的比较。通过使用内部控制,这种偏差被消除,因为对照和测试基因组窗口将具有相同水平的丢弃率。
[0164]
loh阈值和loh调用
[0165]
可选地,可以对从先前步骤获得的loh评分进行阈值化,以定义loh中的基因组区域。在大多数情况下,在基因组窗口中检测到的具有恒定数量基因座的杂合基因座的数量和比例会随着读段深度的增加而增加。为了允许将loh评分阈值化为预先计算的值,每个样本中的映射读段数量优选地标准化为固定读段数量。这种标准化是通过随机采样读段,映射到参考基因组,直到达到所需的次数来执行的(优选包含在1000000次映射读段到10000000个映射读段的范围内)。当通过对“内部”对照执行统计检验来计算loh评分时,上述考虑则不适用。
[0166]
优选地,在loh评分计算为杂合基因座数量的情况下,数据首先被下采样到1000000次映射读段。使用检测到的固定数量(例如n=500;n=1000;n=1500)基因座的窗口对由至少1个读段覆盖的基因座进行分区。一些优选的阈值分别是500、1000和1500个基因座中的3、6、9个杂合snp(图12)。如果loh评分低于所选阈值,则在给定的基因组窗口中调用loh。
[0167]
更详细地说,图12示出了用于定义loh评分阈值的roc分析,loh评分阈值被定义为被至少1个读段与1000000个映射读段所覆盖的(a)n=500,(b)n=1000,(c)n=1500个snp的窗口中的双等位基因snp的数量。使用通过高通全基因组测序和b等位基因频率分析在肿瘤细胞中检测的loh作为参考。
[0168]
在loh评分计算为p值的情况下,由于应用了统计检验,得到一些优选的阈值可以是例如5*10-2或1*10-2。如果loh评分低于所选阈值,则在基因组窗口中调用loh。
[0169]
一旦loh评分达到阈值,loh状态就可以根据以下描述的不同标准指派给基因组区域。
[0170]
1)通过合并窗口的loh区域调用。在该优选实施方式中,如果包含在基因组区域中的每个基因组窗口的loh评分通过阈值化步骤,则将loh状态指派给该基因组区域。
[0171]
2)根据基因组窗口中loh状态的loh区域调用。在该优选实施例中,如果基因组区域中包含的基因组窗口的给定百分比/部分通过阈值化步骤,则将loh状态指派给该基因组区域。例如,如果基因组区域中超过66%、75%、80%、85%、90%、95%的窗口通过阈值化步骤,则将loh状态指派给该基因组区域。
[0172]
3)包含肿瘤抑制基因的基因组区域中的loh调用。在该优选实施方式中,至少一个基因组区域包含肿瘤抑制基因。
[0173]
优选地,所述基因选自brca1、brca2、palb2、tp53、cdkn2a、rb1、apc、pten、cdkn1b、dmp1、nf1、aml1、egr1、tgfbr1、tgfbr2和smad4。
[0174]
样本纯度
[0175]
可以在源自不同种类细胞(例如:肿瘤细胞和正常细胞)混合物的dna中鉴定loh。样本纯度定义为混合物中属于目的类型(例如:肿瘤细胞)的样本的百分比。
[0176]
例如,当克隆性(clonal)#tc肿瘤细胞(即基因组相同,因此具有相同的loh和cna模式)与来自同一个体的#nc正常细胞混合时,所得样本的纯度将是#tc/(#tc+#nc),并且在整个基因组中是均匀的。
[0177]
概括地说,这里的纯度是指与包含一个或多个基因组区域的给定目的区域中的loh状态相关的概念。目的区域可能与整个参考基因组一样大(如上例所示)或小至100kbp。
[0178]
例如,在代表源自同一最后共同祖先肿瘤细胞的不同克隆的肿瘤细胞池存在的情况下,当基因组区域loh状态在源自最后共同祖先的所有克隆中普遍存在时,不同基因组区域的纯度可能会有所不同,从最小的1/池中细胞数到(当loh区域仅在一个细胞中表示时)到最大100%。
[0179]
分析loh的样本优选具有至少50%、更优选至少70%的纯度,这从13可以理解,图13示出了在不同映射读段(1000000-10000000个读段)和样本纯度(10%-90%)下loh评分的接受者操作特征(roc)曲线下面积(auc)值。loh评分定义为被至少2个读段覆盖的n=150个snp的窗口中杂合snp的数量。通过混合从对相当于靶标纯度的比例(肿瘤:正常)的肿瘤细胞和正常细胞的分析中获得的计算机读段,获得不同纯度的样本。通过高通全基因组测序在肿瘤细胞中检测到的lohs用作参考。
[0180]
大小选择对loh检测的影响
[0181]
如前所述,大小选择优选在制备大规模平行测序文库的步骤c期间或之后进行。可以根据不同的标准选择片段的大小。可以通过不同的标准选择测序方法,这也取决于片段
大小。一般来说,有助于loh分析的基因座(多态或杂合)的数量越高,分辨率(每百万个读段)就越好。
[0182]
图14示出了通过从获得自增加fcenter的真实单细胞样本(使用ampli1用于illumina的lowpass制备的测序文库)的数据中计算机选择测序片段子集而获得的数据。图14a示出了大小选择(带宽100)对相对于片段平均长度(有250000个读段)的drs-wga片段覆盖率的影响;图14b显示了大小选择(带宽100)对碱基对方面的分辨率的影响(至少2个读段覆盖的150个snp的窗口),其中有250000个读段;图14c显示大小选择带宽对固定片段平均长度(500bp)(有250000个读段)下的drs-wga片段覆盖率的影响;图14d显示了大小选择带宽对固定片段平均长度(500bp)(有250000个读段)下的分辨率(bp)的影响;图14e显示了读段数量对固定片段平均长度(500bp)下的drs-wga片段覆盖率的影响。至少2个读段覆盖的片段和覆盖的片段总数的比例与映射读段的数量成比例增加(虚线);图14f显示了读段数量对固定片段平均长度(500bp)下分辨率(bp)的影响。
[0183]
这些数据表明,drs-wga片段的总数减少,同时对调用snp有用的被多于一个读段覆盖的片段数量增加,在500bp时达到平台期(图14a)。分辨率由此增加,如具有固定数量snp的基因组窗口的长度减少所示(n=150;图14b)。当不同的带宽应用于给定的映射读段数量和fcenter时,片段的覆盖率和分辨率随着带宽的降低而增加(图14c和14d)。分辨率也随着映射读段数量增加(图14e和14f)。
[0184]
实施例
[0185]
下表3总结了在以下公开的3个实施例中使用的方法的特征。
[0186]
表3
[0187][0188]
实施例1
[0189]
在实施例1中,考虑了从受多发性骨髓瘤影响的男性患者获得的1个循环肿瘤细胞(ctc;测试)和1个白细胞(wbc;对照)的用于illumina dna文库的ampli1 lowpass。测序读段被映射到hg19参考人类基因组,并以1、2、3、4、5、6、7、8、9百万个读段进行下采样。从两个文库中提取dbsnp多态基因座处的等位基因(次要等位基因频率≥5%的dbsnp150常见变体)。用固定的10000000bp基因组窗口分割基因座。采用单侧fisher精确检验来评估两种分类之间关联的显著性(表4),零假设是wbc(对照)和ctc(检验)中杂合基因座和纯合基因座很可能相同。
[0190]
表4
[0191][0192]
每个下采样级别的测试结果如图15所示。从200万个读段开始,该方法在检测11号和13号染色体上的已知loh事件方面表现出高灵敏度。
[0193]
详细地,图15在顶部图显示了来自患有多发性骨髓瘤的患者的ctc的拷贝数图。x轴是染色体;y轴是拷贝数。每个点代表固定大小的基因组窗口。拷贝数区段用实线表示。参考(ref)轨迹显示在拷贝数图下方,它表示通过相同ctc的高通全基因组测序检测到的已知loh区域,以纯黑色显示。下方是标记为1m到9m的轨迹:不同数量的读段(1到9百万)下fisher精确检验结果的对数p值(底数=10)热图。更显著的值由更深的灰色阴影表示。
[0194]
实施例2
[0195]
在实施例2中,使用实施例1中使用的相同单个ctc数据作为输入,并在100万个读段时对数据进行下采样。在这种情况下,基因座在窗口中被分区为至少有1个读段覆盖的固定数量(n=1000)的基因座。对于loh区域的识别,loh评分计算为每个窗口中杂合位置的数量。
[0196]
图16示出了通过使用具有恒定数量的基因座的基因组窗口来检测loh。特别是,顶部图显示了与实施例1相同的ctc的拷贝数图。x轴是染色体;y轴是拷贝数。每个点代表固定大小的基因组窗口。拷贝数区段用实线表示。图下方是热图,表示每个基因组窗口的杂合计数。loh评分更低(杂合基因座数量更少)的窗口更可能处于loh状态,由更深的灰色阴影表示。11号染色体、13号染色体的大臂和x染色体(在男性个体中为单拷贝)显示较低的loh评分。
[0197]
为了确定在loh状态下调用基因组窗口的loh评分阈值,使用与测试样本相同的方法(1000000个映射读段和n=1000个snp窗口)分析了具有已知loh区域的9个单细胞的训练集。然后进行roc分析,最大loh评分阈值=6被确定为灵敏度和特异性之间的最佳折衷点(图17,其中x轴表示1-特异性(值越低表示检测越有特异性)和y轴代表灵敏度。使用通过高
通全基因组测序在肿瘤细胞中检测到的loh作为参考)。
[0198]
该方法成功识别了11号和13号染色体上的loh事件。loh状态也被分配给染色体x,正如在基因组包含单个拷贝的x染色体的男性个体中预期的那样(图18-loh评分低于固定阈值(《=6)且大于10000000bp的区域以黑色显示)。
[0199]
实施例3
[0200]
在实施例3中,分析了从男性患者的经典霍奇金淋巴瘤样本的ffpe组织中获得的2个单个霍奇金里德/斯腾伯格(hodgkin reed/sternberg,hrs)细胞的用于illumina文库的ampli1 lowpass。两个hrs细胞共享相同的拷贝数谱。将测序读段映射到hg19参考人类基因组,并从两个文库中提取dbsnp多态基因座处存在的等位基因(次要等位基因频率≥5%的dbsnp150常见变体)。使用通过使用control-freec软件获得的拷贝数区段对基因座分区,实现基于gc的标准化和拷贝数信号分割[boeva,v等人,生物信息学(bioinformatics),27(2),268-269.http://doi.org/10.1093/bioinformatics/btq635)。使用由拷贝数等于细胞倍性(拷贝数=2)的所有区域联合定义的内部对照。对于通过拷贝数分析定义并包含在染色体臂中的每个区段,进行单侧fisher精确检验以拒绝零假设,该零假设即观察到的双等位基因和单等位基因基因座在区段中和在内部对照中很可能相同(图19-顶部图:一个代表性hrs细胞拷贝数谱。下部图:作为fisher检验的输出获得的p值的-log10的热图。仅显示p值≤0.01的基因组区域。更重要的值由更深的灰色阴影表示)。正如预期的那样,所有拷贝数=1的区域都被正确检测为loh基因组区域。尽管拷贝数=2,但在loh状态下检测到染色体x的长臂。这是意料之中的,因为样本来自男性个体,因此基因组包含单个x染色体。此外,在loh中调用了9q号染色体,仅使用拷贝数信息(拷贝数=2)会遗漏。
[0201]
优点
[0202]
根据本发明的方法适用于分析从测试样本的基因组dna的低通测序获得的数据以检测loh事件。与将loh推断为连续纯合基因座的运行,并需要在一定数量的基因座处提取真实基因型的其他方法相反,本发明的方法基于以下原理:通过分析包含足够数量的低覆盖率测序的基因座的基因组窗口,并通过提取在所述基因座处观察到的等位基因(不一定代表样本基因型),与通过分析正常二倍体样本观察到的情况相比,可以将loh事件检测为双等位基因基因座的减少。
[0203]
与从替代等位基因频率(b等位基因频率或baf)推断loh,要求基因组的高覆盖率例如30x(boeva等人,生物信息学,vol.28no.3(2012),第423-42页)的其他方法相反,根据本发明的方法与低通全基因组测序数据(《1x或更低,低至例如0.05x或甚至0.01x)一起工作,具有相应的成本节约。
[0204]
根据本发明的用于从样本分析loh的方法允许使用非常少的样本而从低通全基因组测序数据到单细胞分辨率推断至整个基因组的loh区域,因为可能在样本非常少的情况下,只有少数(低至一个)ctc可用,额外的可选可能性是在没有正常对照的情况下运行分析,并且读段数量相对较少。
[0205]
此外,该方法的特定实施方式能够通过在文库制备过程中引入某些处理步骤来提高loh调用的分辨率,而不会增加测序成本。
[0206]
根据本发明的方法令人惊讶地以本领域技术人员以前认为无法实现的性能改进了现有技术。特别地,该方法允许:
[0207]-通过低通全基因组测序识别单细胞上的loh,平均覆盖率低至0.01-0.04(人类基因组的250000至1000000个单端150bp读段);
[0208]-在没有对照样本的情况下获得上述要点;
[0209]-获得上述要点,及进一步获得用于研究所述单细胞的其他特征的额外遗传材料的可能性,以及可靠地重新分析单细胞以用于验证的可能性,这是由于在过程中使用了固有的wga。
[0210]
此外,根据本发明的方法甚至允许从微量细胞、ffpe或组织活检确定全基因组拷贝数谱和loh。
[0211]
根据意大利知识产权法典第170条之二(2)的声明
[0212]
本发明中使用的人类来源的生物材料是依据适用的法律规定而获得的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1