用于烯烃聚合的齐格勒纳塔催化剂的制作方法
1.本发明涉及可选单取代的2,2-二(四氢呋喃基)甲烷,更具体地涉及它们在齐格勒-纳塔催化剂中用作内部给体以获得具有所需性质(特别是在分子量分布(mwd)和化学组成分布(ccd)方面)的聚合物的用途。本发明进一步涉及包含所述可选单取代的2,2-二(四氢呋喃基)甲烷的齐格勒-纳塔催化剂组分和用于烯烃聚合的包含所述齐格勒-纳塔催化剂组分的齐格勒-纳塔催化剂,以及制备它们的方法及它们在提供聚烯烃中的用途。
背景技术:
2.齐格勒-纳塔(zn)型聚烯烃催化剂在生产聚烯烃例如乙烯(共)聚合物的领域中是众所周知的。通常,催化剂至少包含由如下形成的催化剂组分:元素周期表(iupac,无机化学命名法,2005)第4至6族的过渡金属化合物、元素周期表(iupac)第1至3族的金属化合物、可选的元素周期表(iupac)第13族元素的化合物和可选的内部电子给体。zn催化剂还可以包含另外的催化剂组分,例如助催化剂和可选的外部电子给体。
3.内部电子给体和内部给体具有相同的含义并且在本发明中可以互换。同样地,这也分别适用于外部电子给体和外部给体。
4.用于生产聚烯烃(例如乙烯(共)聚合物)的催化剂组合物决定了聚合物的性质。因此,催化剂组合物允许“定制”所生产聚合物的性能。
5.ep 373999 a1公开了使用齐格勒-纳塔催化剂来控制聚乙烯均聚物和共聚物的分子量分布(mwd)的方法,该齐格勒-纳塔催化剂包含选自单醚(例如四氢呋喃)的外部电子给体。建议在助催化剂存在的情况下将单醚加入催化组分中,但强烈不建议在没有助催化剂的情况下使单醚和催化组分接触。
6.wo 2007051607 a1公开了另一种定制多峰乙烯聚合物性能的可能方案。使用烷基醚型内部电子给体(优选四氢呋喃)以改性齐格勒-纳塔催化剂组分并影响较高分子量(hmw)组分的分子量分布(mwd)。
7.wo 2004055065 a1公开了用于制备乙烯与α-烯烃的共聚物的固体催化剂组分,该固体催化剂组分包含特定摩尔比的ti、mg、卤素和电子给体,其中所述α-烯烃沿聚合物链均匀分布。电子给体(ed)优选为醚,例如四氢呋喃。所述催化剂组分与烷基铝化合物以及可选的外部电子给体一起用于聚合工艺。可选的外部电子给体可以等于或不同于催化剂组分中使用的ed。
8.wo 2007147715 a1公开了用于烯烃聚合的催化剂组分,该催化剂组分通过特定工艺获得,包含mg、ti、卤素和作为内部电子给体的1,3-二醚。使用催化剂与作为内部电子给体的9,9-双(甲氧基甲基)芴来进行丙烯聚合。
9.ep 0376936 a2公开了一种mgcl2负载的齐格勒-纳塔催化剂,其中喷雾干燥的mgcl2/醇载体材料用第ia至iiia族(根据元素周期表(iupac,无机化学命名法,2005)的第1、2和13族)的化合物处理,然后在可选的内部电子给体的存在下用钛化合物钛化。可选的内部给体化合物(在使用内部电子的实施例中为thf或邻苯二甲酸二异丁酯)与ticl4一起
添加或在添加ticl4之后添加。然而,ep 0376936 a2的给体改性催化剂的活性远低于先前没有给体的催化剂。此外,在给体处理步骤中,使用了10wt%的三乙基铝溶液和多次催化剂烃洗涤,这导致了大量的有机溶剂的浪费。
10.wo 2014004396 a1公开了催化剂组分,其中使用双杂环化合物作为内部电子给体。催化剂组分用于丙烯聚合。
11.wo 2014096296 a1公开了负载的齐格勒-纳塔催化剂组分,其包含选自双(含氧环)化合物的内部给体,以及此种催化剂组分在制备用于生产高分子量聚乙烯的乙烯聚合中的催化剂体系中的用途。
12.wo 2013098139 a1公开了颗粒状第2族金属/过渡金属烯烃聚合催化剂组分,其通过特定工艺获得,包含1,3-二醚化合物作为内部给体,还公开了此种催化剂组分用于制备烯烃聚合(尤其是丙烯聚合)中使用的催化剂的用途。
13.wo 2016097193 a1公开了用于制备聚烯烃的固体mgcl2系齐格勒-纳塔催化剂组分,其通过如下方法制备:使用第13族元素的化合物和内部有机化合物(双环醚)对mgcl2*mroh加合物进行预处理,进一步用第4至6族过渡金属的化合物进行处理。还公开了所述催化剂组分,以及包含所述固体催化剂组分、作为助催化剂的第13族元素化合物和可选的外部添加剂的齐格勒-纳塔催化剂的制备。
14.尽管在齐格勒-纳塔催化剂制备方面已经取得了很大进展,但仍有进一步改进的空间。此外,如今健康、安全和环境政策是催化剂和其他聚合物生产中的重要因素。换言之,聚合物必须满足某些国家和国际机构对健康和环境的严格要求。被认为是潜在有害化合物的一类物质是邻苯二甲酸盐,其通常用作齐格勒-纳塔型催化剂中的内部电子给体。此外,四氢呋喃也已被列为有害物质。
15.由于这些原因,仍然期望找到其他用于齐格勒-纳塔催化剂的内部给体,其不包含邻苯二甲酸酯和/或四氢呋喃但仍能提供所需聚合物性能(例如高分子量)。此外,从商业角度考虑,此种催化剂应该表现出可再现的形态、组成和性能。
16.还需要找到能够生产具有较宽熔体流动速率(mfr)和密度窗口的共聚物的催化剂,使得可以生产具有窄的mwd(分子量分布)和高共聚单体含量并且低熔融温度的高分子量共聚物。
17.最后,催化剂应显示出一定水平的生产率,使其在生产具有广泛分子量范围的聚合物的商业聚合工艺中可用。
18.基于现有技术的教导,给体改性可能可以改善某些性能。然而,这些改进通常是以牺牲催化剂活性和共聚单体响应为代价来实现的。特别地,通过沉淀法制备的mgcl2系催化剂通常对制备条件的变化很敏感。
技术实现要素:
19.本发明的一个目的是提供内部给体,特别是提供包含所述内部给体的齐格勒-纳塔催化剂组分,其克服了上述缺点、具有环境可持续性并且支持具有所需分子量分布(mwd)和化学组成分布(ccd)的乙烯(共)聚合物的制备。
20.本发明的目的通过使用式(i)的化合物作为内部给体、包含所述内部给体的齐格勒-纳塔催化剂组分、包含齐格勒-纳塔催化剂组分的齐格勒-纳塔催化剂以及它们在烯烃
聚合中的用途来实现,其特征于独立权利要求中所述。从属权利要求涵盖了本发明的优选实施方式。
附图说明
21.本发明的优点体现在实验部分和附图中:
22.图1显示了发明例(ie1至ie4)和比较例(ce1至ce9)的多分散性指数(pdi)与分子量的关系。
23.图2显示了发明例(ie1至ie4)和比较例(ce1至ce9)的熔融温度与共聚单体含量的关系。
24.图3显示了发明例(ie1至ie4)和比较例(ce1至ce9)的活性水平与分子量的关系。
具体实施方式
25.出乎意料地,现已发现如在此和下文讨论地在齐格勒-纳塔催化剂体系中使用式(i)的化合物,在用于乙烯的(共)聚合时提供了优异的活性-氢响应平衡。此外,其使用提供了具有所需分子量、分子量分布(mwd)和化学组成分布(ccd)的乙烯(共)聚合物。
26.(i)内部给体
27.因此,本文提供了式(i)的化合物在齐格勒-纳塔催化剂中作为内部给体的用途,特别是用于乙烯(共)聚合,特别是用于在多阶段工艺中生产乙烯(共)聚合物,
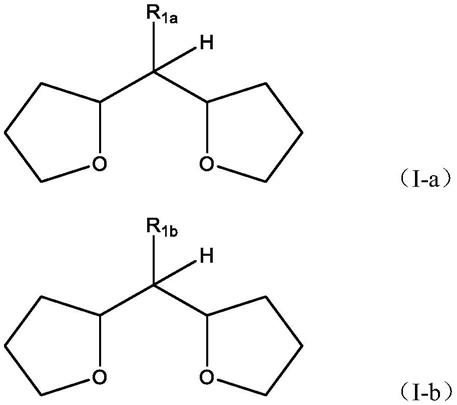
[0028][0029]
式中,
[0030]
r1选自h、直链或支链的c
1-7
烷基、ch2or2和含氧杂环;以及
[0031]
r2选自直链或支链或环状的c
1-8
烷基。
[0032]
优选地,r1选自h、直链或支链的c
1-5
烷基和四氢呋喃基。更优选地,r1选自h、c
1-3
烷基和2-四氢呋喃基。最优选地,r1选自h、甲基和2-四氢呋喃基。
[0033]
从属权利要求和如下说明书中描述了本发明内部给体的用途的优选实施方式。
[0034]
在第一优选实施方式中,式(i)的化合物选自式(i-a)的化合物:
[0035][0036]
式中,
[0037]r1a
选自h、直链或支链的c
1-7
烷基和ch2or2;以及
[0038]
r2选自直链或支链或环状的c
1-8
烷基。
[0039]
优选地,r
1a
选自h和甲基。
[0040]
在第二优选实施方式中,式(i)的化合物选自式(i-b)化合物:
[0041][0042]
式中,
[0043]r1b
选自含氧杂环。
[0044]
优选地,r
1b
选自四氢呋喃基。更优选地,r
1b
为2-四氢呋喃基。
[0045]
在第三优选实施方式中,根据本发明的用途对应于式(i)的化合物作为内部给体在齐格勒-纳塔催化剂中的用途,
[0046][0047]
式中,
[0048]
r1选自h、甲基、ch2or2和含氧杂环;以及
[0049]
r2选自直链或支链或环状的c
1-8
烷基。
[0050]
在第四优选实施方式中,根据本发明的用途对应于式(i)的化合物作为内部给体在齐格勒-纳塔催化剂中的用途,
[0051][0052]
式中,
[0053]
r1选自h、ch2or2和含氧杂环;以及
[0054]
r2选自直链或支链或环状的c
1-8
烷基。
[0055]
术语“含氧杂环”是指含有氧作为具有4至7个环原子的环的一部分的环状结构,其中其他环原子优选为碳。含氧杂环优选为包含1个氧原子作为环成员的5至6元环。含氧杂环可以是饱和的、部分不饱和的、不饱和的或芳族的;优选地,含氧杂环是饱和的。优选地,含氧杂环未被取代。特别地,含氧杂环选自四氢呋喃基;更优选地,含氧杂环为2-四氢呋喃基。
[0056]
在本发明的一个具体实施例中,式(i)的化合物选自二(2-四氢呋喃基)甲烷(dthfm)、1,1-二(2-四氢呋喃基)乙烷(dthfe)和三(四氢呋喃-2-基)甲烷(ffhfm)。在上述化合物中,特别优选dthfm和ffhfm。
[0057]
已出乎意料地发现,通过在齐格勒-纳塔催化剂组分中使用本发明的内部给体,可以生产具有所需分子量、分子量分布(mwd)和化学组成分布(ccd)的多峰聚乙烯(尤其是用于乙烯(共)聚合,特别是用于在多阶段工艺中生产乙烯(共)聚合物)。
[0058]
这些改进,例如分子量的增加和/或mwd的窄化,没有以牺牲催化剂的生产率为代价。相反,生产率保持在可接受的水平。
[0059]
(a)齐格勒-纳塔催化剂组分
[0060]
本文提供了用于烯烃聚合(尤其是乙烯(共)聚合)、特别是用于在多阶段工艺中生产乙烯(共)聚合物的齐格勒-纳塔催化剂组分,所述齐格勒-纳塔催化剂组分包含内部给体,所述内部给体选自式(i)的化合物:
[0061][0062]
式中,
[0063]
r1选自h、直链或支链的c
1-7
烷基、ch2or2和含氧杂环;以及
[0064]
r2选自直链或支链或环状的c
1-8
烷基。
[0065]
优选地,r1选自h、直链或支链的c
1-5
烷基和四氢呋喃基。更优选地,r1选自h、c
1-3
烷基和2-四氢呋喃基。进一步更优选地,r1选自h、甲基和2-四氢呋喃基。
[0066]
从属权利要求和如下说明书中描述了本发明的齐格勒-纳塔催化剂组分的优选实施方式。
[0067]
在齐格勒-纳塔催化剂组分的第一实施例中,式(i)的化合物选自式(i-a)的化合物:
[0068]
式中,
[0069]r1a
选自h、c
1-3
烷基和ch2or2;以及
[0070]
r2选自直链或支链或环状的c
1-8
烷基。
[0071]
优选地,r
1a
选自h和甲基。
[0072]
在齐格勒-纳塔催化剂组分的第二实施例中,式(i)的化合物选自式(i-b)化合物:
[0073]
式中,
[0074]r1b
选自含氧杂环。
[0075]
优选地,r
1b
选自四氢呋喃基。更优选地,r
1b
为2-四氢呋喃基。
[0076]
在齐格勒-纳塔催化剂组分的第三实施例中,式(i)为
[0077][0078]
式中,
[0079]
r1选自h、甲基、ch2or2和含氧杂环;以及
[0080]
r2选自直链或支链或环状的c
1-8
烷基。
[0081]
在齐格勒-纳塔催化剂组分的第四实施例中,式(i)的化合物为
[0082][0083]
式中,
[0084]
r1选自h、ch2or2和含氧杂环;以及
[0085]
r2选自直链或支链或环状的c
1-8
烷基。
[0086]
在一个具体实施例中,式(i)的化合物选自二(2-四氢呋喃基)甲烷(dthfm)、1,1-二(2-四氢呋喃基)乙烷(dthfe)和三(四氢呋喃-2-基)甲烷(ffhfm)。在上述化合物中,特别优选dthfm和ffhfm。
[0087]
本文还提供了如下所述的齐格勒-纳塔催化剂组分。
[0088]
如本文所用,术语“齐格勒-纳塔催化剂组分”是指齐格勒-纳塔催化剂的催化剂组分,也称为主催化剂。本发明的齐格勒-纳塔催化剂组分至少包含过渡金属化合物和内部给体。
[0089]
特别地,术语“齐格勒-纳塔催化剂组分”是指由如下形成的催化剂组分:
[0090]
(ii)元素周期表(iupac,无机化学命名法,2005)第4至6族过渡金属的化合物,
[0091]
(iii)元素周期表(iupac,2005)第1至3族金属的化合物,
[0092]
(iv)可选的元素周期表(iupac,2005)第13或14族元素的化合物;以及
[0093]
(i)如本文所定义的内部给体。
[0094]
(i)内部给体
[0095]
如在此和下文所用,术语“内部给体”,也称为“内部电子给体”,是指作为齐格勒-纳塔催化剂组分的一部分的化合物,即在齐格勒-纳塔催化剂组分的合成过程中添加并且通常作为齐格勒-纳塔催化剂体系中的电子给体。
[0096]
内部给体是齐格勒-纳塔催化剂组分的一部分并且在其合成过程中被加入所述齐
格勒-纳塔催化剂组分中。
[0097]
内部给体(id)与(iii)第1至3族金属化合物的加样(合成期间)摩尔比、特别是内部给体(id)与mg的加样(合成期间)摩尔比(id/mg)优选为0.010至0.300mol/mol,更优选为0.030至0.250mol/mol,甚至更优选为0.040至0.120mol/mol。
[0098]
齐格勒-纳塔催化剂组分的其他化合物
[0099]
除内部给体(i)之外,齐格勒-纳塔催化剂组分还包含:
[0100]
(ii)元素周期表(iupac,无机化学命名法,2005)第4至6族过渡金属的化合物;
[0101]
(iii)元素周期表(iupac,2005)第1至3族金属的化合物;以及
[0102]
(iv)可选的元素周期表(iupac,2005)第13族元素的化合物。
[0103]
齐格勒-纳塔催化剂组分通常是固体。可以在不使用任何外部载体材料的情况下形成固体齐格勒-纳塔催化剂组分,或者它可以是负载在外部载体材料上的固体齐格勒-纳塔催化剂组分。
[0104]
本发明的固体负载的齐格勒-纳塔催化剂组分包含:
[0105]
(i)内部给体,所述内部给体选自如本文所讨论的式(i)的化合物;
[0106]
(ii)元素周期表(iupac,无机化学命名法,2005)第4至6族过渡金属的化合物,特别是钛化合物;
[0107]
(iii)元素周期表(iupac,2005)第1至3族金属的化合物,特别是镁化合物;以及
[0108]
(iv)可选的元素周期表(iupac,2005)第13族元素的化合物,特别是铝化合物;
[0109]
其中,当存在组分(i)至(iv)时,组分(i)至(iv)被负载在固体载体上。
[0110]
固体载体可以选自无机氧化物固体载体,例如二氧化硅、三氧化二铝、二氧化钛、二氧化硅-三氧化二铝、二氧化硅-二氧化钛和mg系固体载体,例如mgcl2系固体载体。优选地,固体载体为二氧化硅或mg系固体载体,例如mgcl2系固体载体。更优选地,固体载体为mgcl2*mroh加合物的固体载体颗粒,其中,mgcl2*mroh加合物中的r为直链或支链的c
1-12
烷基,m为0至6、优选为1至6、更优选为1至4。
[0111]
二氧化硅载体的基于体积的中值粒径(d
v0.5
)通常为2至500μm,优选为5至200μm,更优选为10至100μm。不过,发现如果载体的d
v0.5
粒径为5至30μm、优选7至20μm、更优选8至15μm,则可以获得特殊的益处。或者,二氧化硅载体的d
v0.5
粒径可以为20至80μm,优选为20至30μm。例如,合适的载体材料的实例包括由ineos silicas(前crossfield)生产和销售的es747jr,以及由grace生产和销售的sp9-491。
[0112]
例如,如在ep 0688794 a1和wo 99/51646 a1中描述,催化剂组分可以通过使载体与上述化合物顺序接触来制备。或者,如wo 01/55230 a1中所述,可通过先由组分制备溶液、然后使该溶液与载体接触来制备。
[0113]
或者,本发明中使用的催化剂组分可以负载在mg系固体载体(特别是mgcl2)上。因此,催化剂包含钛化合物和可选的第13族元素化合物(例如铝化合物、二卤化镁,例如二氯化镁)。一组此类齐格勒-纳塔催化剂组分包含钛化合物以及充当载体的卤化镁化合物。此类催化剂公开于例如wo 2005/118655 a1、ep0810235 a2、wo 2014/096296 a1和wo 2016/097193 a1中。
[0114]
例如,催化剂可以如下制备:在催化剂合成开始时,即在用钛化合物(例如ticl4)处理之前或甚至在用第13族元素化合物处理mgcl2*metoh载体材料之前,使球状或粒状的
mgcl2*mroh(例如mgcl2*metoh)载体材料与选自式(i)化合物的内部电子给体接触,最终提取固体催化剂组分。
[0115]
mg系固体载体的中值粒径d
v0.5
通常为2至500μm,优选为5至200μm,更优选为10至100μm。不过,发现如果载体的d
v0.5
粒径为5至30μm、优选为7至25μm、更优选为8至20μm或甚至8至15μm,则可以获得特殊的益处。或者,载体的d
v0.5
粒径可以为20至80μm,优选为20至30μm。mgcl2*mroh可以通过现有技术中描述的方法制备。mgcl2*mroh载体的制备方法描述于多个专利(例如ep 0376936 b1、ep0 424049b1、ep 0655073 b1、us 4071674和ep 0614467 b1,其通过引用并入本文)中。
[0116]
或者,固体齐格勒-纳塔催化剂组分可以在不使用其上负载有活性催化剂组分的任何外部载体材料(例如二氧化硅、三氧化二铝或单独制备的镁系固体载体)的情况下形成。取而代之,固体催化剂通过如下方法形成:使所有活性催化剂化合物以液体形式彼此接触和/或反应,然后形成固体催化剂。
[0117]
在本发明的一个实施方式中,齐格勒-纳塔催化剂组分包含:
[0118]
(i)内部给体,所述内部给体选自如本文所讨论的式(i)的化合物;
[0119]
(ii)第4至6族金属、优选第4族金属、更优选ti,其含量(通过icp分析测定)为齐格勒-纳塔催化剂组分总重量的1.0wt%至15.0wt%、优选2.5wt%至12.0wt%、更优选3.5wt%至9.5wt%;
[0120]
(iii)第1至3族、优选第2族金属、更优选mg,其含量(通过icp分析测定)为齐格勒-纳塔催化剂组分总重量的5.0wt%至30.0wt%、优选9.0wt%至22.0wt%、更优选12.5wt%至18.5wt%;
[0121]
(iv)al,其含量(通过icp分析测定)为齐格勒-纳塔催化剂组分总重量的0.0wt%至3.0wt%、优选0.0wt%至2.6wt%、更优选0.0wt%至1.8wt%。
[0122]
此外,根据本发明的一个实施方式的齐格勒-纳塔催化剂组分的中值粒径d
v0.5
为2至100μm,优选为5至30μm,更优选为8至20μm,甚至更优选为8至15μm。
[0123]
(ii)第4至6族过渡金属化合物
[0124]
元素周期表(iupac,无机化学命名法,2005)第4至6族过渡金属的化合物优选为元素周期表(iupac,2005)第4族过渡金属化合物或钒化合物,更优选为钛化合物或钒化合物,甚至更优选为钛化合物,进一步更优选为含卤素的钛化合物,最优选为含氯的钛化合物。
[0125]
在一个特别优选的实施方式中,钛化合物是式halyti(oalk)
4-y
的含卤素的钛化合物,其中,alk是c
1-20
烷基,优选c
2-10
烷基,更优选c
2-8
烷基;hal为卤素,优选氯;y为1、2、3或4,优选为3或4,更优选为4。
[0126]
合适的钛化合物包括三烷氧基单氯化钛、二烷氧基二氯化钛、烷氧基三氯化钛和四氯化钛。优选地,使用四氯化钛。
[0127]
齐格勒-纳塔催化剂组分中的第4至6族化合物的量优选使得:第4至6族金属(优选第4族金属,更优选ti)的含量(通过icp分析测定)为齐格勒-纳塔催化剂组分总重量的1.0wt%至15.0wt%、优选2.5wt%至12.0wt%、更优选3.5wt%至9.5wt%。如实验部分中所述,第4至6族金属化合物的量通过icp分析测定。
[0128]
(iii)第1至3族金属化合物
[0129]
第1至3族金属的化合物优选为元素周期表(iupac,无机化学命名法,2005)第2族
金属的化合物,更优选为镁化合物。
[0130]
齐格勒-纳塔催化剂组分中的第1至3族的金属化合物的量优选使得:第1至3族金属(优选第2族金属,更优选mg)的含量为齐格勒-纳塔催化剂组分总重量的5.0wt%至30.0wt%、优选9.0wt%至22.0wt%、更优选12.5wt%至18.5wt%。如实验部分中所述,第1至3族的金属化合物的量通过icp分析测定。
[0131]
在一个具体实施例中,第1至3族金属化合物以mgcl2系固体载体的形式提供,优选以mgcl2*mroh加合物的固体载体颗粒的形式提供,其中加合物mgcl2*mroh中的r是直链或支链的c
1-12
烷基,m为0至6、优选为1至6、更优选为1至4。
[0132]
优选地,最终的固体齐格勒-纳塔催化剂组分颗粒的中值粒径d
v0.5
为2至100μm,优选为5至30μm,更优选为8至20μm,甚至更优选为8至15μm。
[0133]
(iv)第13族元素的化合物
[0134]
可选的元素周期表(iupac,无机化学命名法,2005)第13族元素的化合物优选为铝化合物。
[0135]
特别优选地,铝化合物是式al(烷基)
x
hal
3-x
(ii)的铝化合物,其中,每个烷基独立地为c
1-12
烷基,优选c
1-8
烷基,更优选c
1-6
烷基;hal为卤素,优选氯;1<x≤3。烷基可以为直链、支链或环状烷基,或它们的混合物。
[0136]
优选的铝化合物是烷基二氯化铝、二烷基氯化铝或三烷基铝化合物,例如二甲基氯化铝、二乙基氯化铝、二异丁基氯化铝和三乙基铝或它们的混合物。最优选地,铝化合物是三烷基铝化合物,尤其是三乙基铝。
[0137]
齐格勒-纳塔催化剂组分中的第13族元素的化合物的量优选使得:第13族元素(优选al)的含量为齐格勒-纳塔催化剂组分总重量的0wt%至3.0wt%、优选0wt%至2.6wt%、更优选0wt%至1.8wt%。如实验部分中所述,第13族元素的化合物的量通过icp分析测定。
[0138]
内部给体是齐格勒-纳塔催化剂组分的一部分,并且在所述齐格勒-纳塔催化剂组分的合成过程中被加入所述齐格勒-纳塔催化剂组分中。
[0139]
内部给体(id)与(iii)第1至3族金属(特别是mg)的加样摩尔比(合成中)(id/mg)优选为0.010至0.300mol/mol,更优选为0.030至0.250mol/mol,甚至更优选为0.040至0.120mol/mol。
[0140]
最终齐格勒-纳塔催化剂组分通常具有:
[0141]
mg/ti mol/mol比率为2.0至15.0,优选为2.5至12.0,更优选为2.8至7.0;
[0142]
al/ti mol/mol比率为0至1.0,优选为0至0.8,更优选为0至0.5;以及
[0143]
cl/ti mol/mol比率为5.0至30.0,优选为6.0至27.0,更优选为9.0至16.0。
[0144]
齐格勒-纳塔催化剂组分的制备方法
[0145]
通常,本文所定义的本发明齐格勒-纳塔催化剂组分通过将本发明的内部给体(即式(i)化合物)加入到齐格勒-纳塔催化剂组分的制备方法中来制备。这可以通过制造齐格勒-纳塔催化剂领域的技术人员熟知的方式和条件来完成。
[0146]
因此,本文提供了一种用于制备齐格勒-纳塔催化剂组分(特别是如本文所定义的齐格勒-纳塔催化剂组分)的方法,该方法包括将本文所定义的式(i)的内部给体加入到制备齐格勒-纳塔催化剂组分的方法中。
[0147]
如ep0376936a1、ep0688794 a1或wo99/51646 a1中所述,固体齐格勒-纳塔催化剂
组分可以例如通过使固体载体(例如二氧化硅、三氧化二铝或单独制备的mg系固体载体,例如mgcl2系固体载体)与上述化合物顺序接触来制备。或者,如wo 01/55230 a1中所述,它可以通过先由组分制备溶液、然后使该溶液与固体载体接触来制备。
[0148]
在本方法的一个具体实施例中,制备齐格勒-纳塔催化剂组分的方法包括如下步骤:
[0149]
(m-a)提供固体载体、优选mgcl2系固体载体、更优选mgcl2*mroh加合物的固体载体颗粒,其中,mgcl2*mroh加合物中的r为直链或支链的c
1-12
烷基,m为0至6、优选为1至6、更优选为1至4;
[0150]
(m-b)用第13族元素的化合物对步骤(m-a)的固体载体颗粒进行预处理;
[0151]
(m-c)用第4至6族的过渡金属化合物对步骤(m-b)的经预处理的固体载体颗粒进行处理;
[0152]
(m-d)提取齐格勒-纳塔催化剂组分;
[0153]
其中,在步骤(m-c)中处理固体载体之前,使固体载体与式(i)的内部有机化合物或包含其的混合物接触。
[0154]
优选地,在用第13族元素化合物对固体载体进行预处理之前、期间或之后,但在用第4至6族的过渡金属化合物对固体载体进行处理之前,加入如本文所定义的式(i)的化合物。
[0155]
固体齐格勒-纳塔催化剂组分也可以在不使用其上负载有活性催化剂组分的任何外部载体材料(例如二氧化硅、三氧化二铝或单独制备的镁系固体载体)的情况下形成。在此情况下,固体催化剂通过如下方法形成:使所有活性催化剂化合物以液体形式彼此接触和/或反应,然后形成固体催化剂。固体齐格勒-纳塔催化剂组分可以通过乳液-固化法或通过沉淀法形成。齐格勒-纳塔催化剂组分通过乳液-固化法还是通过沉淀法形成取决于条件,特别是取决于使化合物接触期间所使用的温度。在乳液-固化法中,齐格勒-纳塔催化剂组分的化合物在具有至少两个液相的乳液中形成分散相(即不连续相)。液滴形式的分散相从乳液中固化,其中形成固体颗粒形式的齐格勒-纳塔催化剂组分。这些类型催化剂的制备原理已在例如wo 2003/106510 a1、wo 2013/098139 a1和wo 2014/096297 a1中给出。
[0156]
齐格勒-纳塔催化剂
[0157]
因此,本文进一步提供了用于烯烃聚合、特别是乙烯(共)聚合的齐格勒-纳塔催化剂,所述齐格勒-纳塔催化剂包含本文所定义的催化剂组分,其中催化剂组分包含内部给体,所述内部给体选自式(i)的化合物。因此,本文还提供了齐格勒-纳塔催化剂在烯烃聚合、特别是乙烯(共)聚合中的用途,所述齐格勒-纳塔催化剂包含齐格勒-纳塔催化剂组分,所述齐格勒-纳塔催化剂组分包含内部给体,所述内部给体选自式(i)的化合物:
[0158][0159]
式中,
[0160]
r1选自h、直链或支链的c
1-7
烷基、ch2or2和含氧杂环;以及
[0161]
r2选自直链或支链或环状的c
1-8
烷基。
[0162]
优选地,r1选自h、直链或支链的c
1-5
烷基和四氢呋喃基。更优选地,r1选自h、c
1-3
烷基和2-四氢呋喃基。甚至更优选地,r1选自h、甲基和2-四氢呋喃基。
[0163]
优选地,r1选自h、甲基和四氢呋喃基。更优选地,r1选自h、甲基和2-四氢呋喃基。
[0164]
从属权利要求和如下说明书中描述了本发明的齐格勒-纳塔催化剂的用途的优选实施方式。
[0165]
在齐格勒-纳塔催化剂及其用途的第一实施例中,式(i)的化合物选自式(i-a)的化合物:
[0166][0167]
式中,
[0168]r1a
选自h、c
1-3
烷基和ch2or2;以及
[0169]
r2选自直链或支链或环状的c
1-8
烷基。
[0170]
优选地,r
1a
选自h和甲基。
[0171]
在齐格勒-纳塔催化剂的第二实施例中,式(i)的化合物选自式(i-b)化合物:
[0172][0173]
式中,
[0174]r1b
选自含氧杂环。
[0175]
优选地,r
1b
选自四氢呋喃基。更优选地,r
1b
为2-四氢呋喃基。
[0176]
在齐格勒-纳塔催化剂的第三实施例中,式(i)为
[0177][0178]
式中,
[0179]
r1选自h、甲基、ch2or2和含氧杂环;以及
[0180]
r2选自直链或支链或环状的c
1-8
烷基。
[0181]
在齐格勒-纳塔催化剂组分的第四实施例中,式(i)的化合物为
[0182][0183]
式中,
[0184]
r1选自h、ch2or2和含氧杂环;以及
[0185]
r2选自直链或支链或环状的c
1-8
烷基。
[0186]
在一个具体实施例中,式(i)的化合物选自二(2-四氢呋喃基)甲烷(dthfm)、1,1-二(2-四氢呋喃基)乙烷(dthfe)和三(四氢呋喃-2-基)甲烷(ffhfm)。在上述化合物中,特别优选dthfm和ffhfm。
[0187]
还提供了本文所公开的齐格勒-纳塔催化剂。
[0188]
特别地,本发明的齐格勒-纳塔催化剂包含:
[0189]
(a)本文所定义的齐格勒-纳塔催化剂组分;
[0190]
(b)助催化剂,所述助催化剂选自元素周期表(iupac,2005)第13族元素化合物;以及
[0191]
(c)可选的外部给体。
[0192]
(b)助催化剂(ii)
[0193]
齐格勒-纳塔催化剂通常与助催化剂(也称为活化剂)一起使用。合适的助催化剂是元素周期表(iupac,2005)第13族元素的化合物,通常是第13族元素的c
1-16
烷基化合物,尤其是c
1-16
烷基铝化合物。这些化合物包括:三烷基铝化合物,例如三甲基铝、三乙基铝、三异丁基铝、三己基铝和三正辛基铝;烷基铝卤化物,例如乙基二氯化铝、二乙基氯化铝、乙基倍半氯化铝、二甲基氯化铝等。特别优选的活化剂是三烷基铝,其中特别使用三乙基铝、三甲基铝和三异丁基铝。
[0194]
助催化剂的量取决于具体的催化剂和助催化剂。通常,例如三乙基铝的量使得:铝与过渡金属的摩尔比(例如al/ti)为1至1000mol/mol,优选为3至100mol/mol,特别是约5至约30mol/mol。
[0195]
(c)外部给体
[0196]
本发明的催化剂还可以包含外部给体。可以使用的外部给体包括醚化合物,通常是四氢呋喃、硅氧烷或硅烷型外部给体和/或现有技术中已知的烷基卤化物。外部给体也称为外部电子给体。外部电子给体不是固体催化剂组分的一部分,而是作为单独的组分加入聚合工艺中。
[0197]
烯烃聚合
[0198]
本文所定义的本齐格勒-纳塔催化剂组分,特别是本文所定义的齐格勒-纳塔催化剂,旨在用于聚合烯烃(优选乙烯)和可选的c
2-20
共聚单体。
[0199]
因此,本文提供了本文所定义的齐格勒-纳塔催化剂组分或本文所定义的齐格勒-纳塔催化剂在烯烃聚合、优选乙烯(和可选的c
2-20
共聚单体)(共)聚合中的用途。
[0200]
此外,本文提供了烯烃聚合、特别是乙烯(共)聚合的方法,所述方法包括将齐格
勒-纳塔催化剂引入聚合反应器,所述齐格勒-纳塔催化剂包含如本文所限定的齐格勒-纳塔催化剂组分。
[0201]
优选地,本文所定义的齐格勒-纳塔催化剂组分或本文所定义的齐格勒-纳塔催化剂用于乙烯和可选的一种以上共聚单体的(共)聚合。乙烯聚合中常用的共聚单体是α-烯烃共聚单体。α-烯烃共聚单体优选选自c
3-20
α-烯烃,更优选选自c
4-10
α-烯烃(例如1-丁烯、异丁烯、1-戊烯、1-己烯、4-甲基-1-戊烯、1-庚烯、1-辛烯、1-壬烯和1-癸烯)、二烯(例如丁二烯、1,7-辛二烯和1,4-己二烯)或环状烯烃(例如降冰片烯)以及它们的任意混合物。最优选地,共聚单体是1-丁烯和/或1-己烯。
[0202]
本发明的催化剂允许生产范围广泛的聚乙烯(共)聚合物。因此,可以生产高密度、中密度和低密度乙烯(共)聚合物。
[0203]
如果共聚物是所需的最终产品,则乙烯共聚物的共聚单体含量可以根据所需的聚合物性能在宽范围内变化。因此,共聚单体含量可以为0.1wt%到20wt%,优选为0.5wt%至15wt%,更优选为1.0wt%至10wt%。
[0204]
本文进一步提供了生产乙烯均聚物或乙烯共聚物的方法,所述方法包括如下步骤:
[0205]
(p-a)将本文所定义的齐格勒-纳塔催化剂组分引入聚合反应器;
[0206]
(p-b)将能够活化所述主催化剂的助催化剂引入聚合反应器;
[0207]
(p-c)将乙烯、可选的c
3-c
20
α-烯烃共聚单体和可选的氢气引入聚合反应器;以及
[0208]
(p-d)将所述聚合反应器保持在生产乙烯均聚物或乙烯共聚物的此种条件下。
[0209]
可以通过本领域已知的任何方式将催化剂转移到聚合区。因此,可以将催化剂悬浮在稀释剂中并将其保持为均匀浆体。特别优选使用如wo 2006/063771 a1中公开的粘度为20至1500mpa
·
s的油作为稀释剂。还可以将催化剂与油脂和油的粘性组合物混合,将所得糊状物送入聚合区。此外,可以使催化剂沉降,将由此获得的催化剂泥的一部分以例如ep0428054a1中公开的方式引入聚合区。
[0210]
根据本发明使用的聚合工艺包括:至少一个气相反应器,或至少一个浆体反应器,或至少一个浆体反应器和至少一个气相反应器的组合。
[0211]
在浆体中的聚合通常在惰性稀释剂中进行,所述惰性稀释剂通常为烃稀释剂,例如甲烷、乙烷、丙烷、正丁烷、异丁烷、戊烷、己烷、庚烷、辛烷等或它们的混合物。优选地,稀释剂是c
1-4
低沸点烃或此类烃的混合物。特别优选的稀释剂是丙烷,可能含有少量的甲烷、乙烷和/或丁烷。
[0212]
浆体聚合的温度通常为40至115℃,优选为60至110℃,特别是70至100℃。压力为1至150巴,优选为10至100巴。
[0213]
浆体聚合可以在任何已知的用于浆体聚合的反应器中进行。此种反应器包括连续搅拌釜反应器和环路反应器。特别优选在环路反应器中进行聚合。如本领域已知,可选地将氢气进料到反应器中以控制聚合物的分子量。
[0214]
此外,可以将一种以上的α-烯烃共聚单体加入反应器中以控制聚合物产物的密度和形态。此种氢气和共聚单体进料的实际量取决于所得聚合物的所需的熔融指数(或分子量)和密度(或共聚单体含量)。
[0215]
在气相中的聚合可以在流化床反应器、快速流化床反应器或沉降床反应器或它们
的任意组合中进行。
[0216]
通常,流化床或沉降床聚合反应器在50至100℃、优选65至90℃的温度下运行。压力合适地为10至40巴,优选为15至30巴。
[0217]
此外,如果需要,可以将抗静电剂引入浆体和/或气相反应器。
[0218]
该工艺还可包括预反应器和后反应器。
[0219]
聚合步骤之前可以存在预聚合步骤。预聚合步骤可以在浆体或气相中进行。优选地,预聚合在浆体中进行,尤其是在环路反应器中进行。预聚合步骤中的温度通常为0至90℃,优选为20至80℃,更优选为30至70℃。
[0220]
压力不是关键,通常为1至150巴,优选为10至100巴。
[0221]
聚合可以连续或分批地进行,优选聚合连续地进行。
[0222]
用于生产根据本发明的乙烯(共)聚合物的优选的多阶段工艺包括浆体聚合阶段和气相聚合阶段。每个阶段可以包括一个以上的聚合反应器。合适的反应器配置包括一个至两个浆体反应器(优选环路反应器)和一个气相反应器。此种聚合配置描述于例如北欧化工的专利文献例如wo92/12182a1和wo96/18662a1中,称为borstar技术。
[0223]
在本方法的第一个实施例中,聚合烯烃在多阶段聚合工艺中完成,所述多阶段聚合工艺包括至少一个用于生产乙烯(共)聚合物的气相反应器。
[0224]
在本方法的第二个实施例中,烯烃的聚合在多阶段聚合工艺中完成,所述多阶段聚合工艺包括至少一个浆体反应器(优选两个浆体反应器)和一个气相反应器。
[0225]
聚合物特性
[0226]
通过在根据本发明的聚合工艺中使用齐格勒-纳塔催化剂组分中的本发明内部给体,可以生产具有所需的分子量、分子量分布(mwd)和化学组成分布(ccd)的乙烯(共)聚合物,同时保持良好的生产率水平,如下文实施例所示。
[0227]
所生产聚合物的分子量、分子量分布(mwd)和化学组成分布(ccd)可以通过利用齐格勒-纳塔催化剂中的本发明内部给体来优化。
[0228]
此外,改进(例如分子量的增加、化学组成分布的改进和mwd的窄化)没有以牺牲催化剂的生产率为代价,而是生产率保持在可接受的水平。因此,包含本发明的内部给体的齐格勒-纳塔催化剂的性能提供了改进,例如分子量的增加、化学组成分布的改进和mwd的窄化,这些改进没有以牺牲催化剂的生产率为代价,而是生产率保持在可接受的水平。
[0229]
特别地,氢响应、mwd、共聚单体响应、化学组成分布(ccd)、催化剂活性和生产率的最佳组合使得在齐格勒-纳塔催化剂中使用本发明的内部给体对于聚乙烯(共)聚合物的生产非常有吸引力。
[0230]
实验部分
[0231]
分析方法
[0232]
通过icp-oes测定催化剂组分中的al、mg、ti含量
[0233]
将由干燥的催化剂组分粉末组成的样品进行混合,以便取出有代表性的测试部分。在惰性气氛中,将约20至50mg的材料取样到20ml体积的小瓶中,记录粉末的精确重量。
[0234]
如下所述地在容量瓶中制备已知体积(v)的测试溶液。通过加入少量去离子蒸馏(di)水(5%v)在冷却的小瓶中进行样品消解,然后加入浓硝酸(65%hno3,5%v)。将混合物转移到容量瓶中。用di水将溶液稀释至最终体积v,并稳定两小时。
[0235]
使用thermo elemental icap 6300电感耦合等离子体-光学发射光谱仪(icp-oes),在室温下对所得含水样品进行元素分析。该仪器使用空白溶液(5%hno3溶液)和六种标准溶液(在5%hno3去离子水溶液中的0.5ppm、1ppm、10ppm、50ppm、100ppm和300ppm的al、ti和mg)针对al、ti和mg进行校准。校准曲线使用曲线线性拟合和1/浓度加权。
[0236]
在分析之前,立即使用空白溶液和300ppm al、100ppm ti、mg标准溶液对校准进行验证和调整(仪器功能称为“re-slope”)。运行质量控制样品(qc;20ppm al和ti、50ppm mg,在5%hno
3 di水溶液中)以确认“re-slope”。qc样品也在每5个样品之后和计划分析组结束时运行。
[0237]
使用285.213nm线监测镁的含量,使用336.121nm线监测钛的含量。当测试部分中的铝浓度为0至10wt%时,通过167.079nm线监测铝含量;当铝浓度高于10wt%时,通过396.152nm线监测铝含量。
[0238]
报告值是取自同一样品的三个连续等分试样结果的平均值,并根据输入到软件中的测试部分的原始重量和稀释体积将报告值关联至原始催化剂样品。
[0239]
通过电位滴定法测定催化剂组分中的cl含量
[0240]
催化剂组分的氯化物含量通过用硝酸银滴定来测定。在氮气下在隔膜密封的小瓶中称量50至200mg的催化剂组分的测试部分。使用注射器将1份浓hno3(68%,分析级)和4份去离子蒸馏(di)水的溶液以2.5ml的等分试样加入样品中。在反应完成和催化剂组分材料溶解后,使用过量di水将溶液转移到滴定杯中。然后立即在mettler toledo t70自动滴定仪中用商业认证的0.1m agno3溶液滴定该溶液。使用ag电极确定滴定终点。氯化物总量由滴定计算并与原始样品重量相关。
[0241]
通过gc-ms测定催化剂组分中的挥发物
[0242]
通过在水和二氯甲烷中对样品和内标进行液液萃取,使用40至60mg测试部分的催化剂组分粉末制备测试溶液:首先向测试部分中加入10ml二氯甲烷,然后使用精密微量注射器加入1ml内标溶液(庚二酸二甲酯,0.71体积%,在去离子水中)。将悬浮液超声处理30分钟,静置以进行相分离。从有机相中取出一部分测试溶液,使用0.45μm针头式过滤器过滤。
[0243]
对于校准,通过将五份逐渐增加的分析物标准材料准确地加入容量瓶中并用甲醇定容至刻度,来制备具有不同分析物浓度的五种标准储备溶液。为制备校准样品,用与样品相同体积比的istd水溶液和二氯甲烷从储备溶液中萃取出200μl等分试样。最终校准样品中的分析物含量为0.1mg至15mg。
[0244]
使用配备有agilent 5977a质谱检测器的agilent 7890b气相色谱仪进行测量。使用zb-xlb-ht inferno 60m
×
250μm
×
0.25μm色谱柱(phenomenex)通过三通道辅助epc和3m
×
250μm
×
0μm的柱前限制毛细管进行中点反吹,实现分离。初始烘箱温度为50℃,保温时间为2分钟。烘箱温坡由第一阶段(以5℃/min升至150℃)和第二阶段(以30℃/min升至300℃)组成,然后在300℃进行1分钟的后运行反吹。
[0245]
入口以分流模式运行。进样体积为1μl,进样口温度280℃,隔垫吹扫3ml/min,总流速67.875ml/min,分流比50:1。载气为99.9996%he,柱前流速为1.2721ml/min,从反吹epc到分析柱的额外流速为2ml/min。ms检测器传输线保持在300℃。msd在70ev的电子冲击模式和15-300m/z的扫描模式下运行。
[0246]
信号检出由保留时间(庚烷4.8、甲苯6.3、庚二酸二甲酯23.2)和目标离子m/z(庚烷71.1、甲苯91.1、庚二酸二甲酯157.1)确定。此外,使用定性离子确认庚烷、甲苯的检出。对每种分析物和内标的目标离子信号进行积分并与校准曲线进行比较,校准曲线在每次运行开始时用五个校准样品建立。响应比的校准曲线是线性的,没有样品浓度加权。每次运行均使用质量控制样品来验证标准化。所有测试溶液均进行两次重复试验运行。测试部分的质量用于计算两次重复试验的样品中的分析物浓度,将结果报告为平均值。
[0247]
通过dsc测定聚合物熔融和结晶特性
[0248]
使用mettler toledo dsc2对5至10mg样品进行聚合物差示扫描量热分析(dsc)。将聚合物粉末或颗粒切割(pellet cut)或mfr线切割(mfr string cut)样品置于40μl铝盘中,称量精确至0.01mg,用盖子将铝盘密封。dsc根据iso 11357-3或astm d3418在加热/冷却/加热循环中以10℃/min的扫描速率运行。氮气吹扫气体的流量设置为50至80ml/min。第一次加热运行的温度范围为30℃至180℃。冷却运行和第二次加热运行的温度范围为180℃至0℃(或以下)。第一次加热运行和冷却运行的等温时间为5分钟。第一次熔化运行用于去除样品的热历史。结晶温度(tc)由冷却运行确定,而主熔融温度(tm)、结晶度(结晶%)和熔化热(hm)由第二次加热运行确定。
[0249]
聚合物熔体流动速率
[0250]
熔体流动速率根据iso 1133在190℃和给定负载下测量,单位为克/10分钟。熔体流动速率是聚合物分子量的指标。熔体流动速率越高,聚合物的分子量越低。
[0251]
mfr
21
:190℃,21.6kg负载
[0252]
平均分子量和分子量分布(mn、mw、mz、mwd、pdi)
[0253]
根据iso 16014-1:2003、iso 16014-2:2003、iso 16014-4:2003和astm d 6474-12,使用如下公式,通过凝胶渗透色谱法(gpc)来确定平均分子量(mz、mw和mn)、分子量分布(mwd)及其宽度(由多分散性指数pdi=mw/mn描述,其中mn是数均分子量,mw是重均分子量):
[0254][0255][0256][0257]
对于恒定的洗脱体积间隔δvi,其中ai和mi分别为与洗脱体积vi相关的色谱峰切片面积和聚烯烃分子量(mw),其中n等于从积分上下限之间的色谱图中获得的数据点的数量。
[0258]
使用配备有红外(ir)检测器(polymerchar(西班牙巴伦西亚)的ir4或ir5)或差示折光仪(ri,购自安捷伦)的高温gpc仪器,配备有3
×
agilent-plgel olexis色谱柱和1
×
agilent-plgel olexis guard色谱柱。用250mg/l 2,6-二叔丁基-4-甲基苯酚稳定的1,2,4-三氯苯(tcb)被用作溶剂和流动相。色谱系统在160℃和1ml/min的恒定流速下运行。每次分析注入200μl样品溶液。使用agilent cirrus软件3.3版或polymerchar gpc-ir控制软件进行数据收集。
[0259]
使用普适校准(根据iso 16014-2:2003)对色谱柱组进行校准,使用19个窄mwd聚
苯乙烯(ps)标准品,范围为0.5kg/mol至11500kg/mol。ps标准品在室温下溶解数小时。聚苯乙烯峰值分子量到聚烯烃分子量的转换通过使用mark houwink方程和如下mark houwink常数完成:
[0260]kps
=19x 10-3
ml/g,η
ps
=0.655
[0261]kpe
=39x 10-3
ml/g,η
pe
=0.725
[0262]kpp
=19x 10-3
ml/g,η
pp
=0.725
[0263]
三阶多项式拟合被用于拟合校准数据。
[0264]
所有样品均以0.5至1mg/ml的浓度范围制备,在持续温和摇动和160℃下溶解2.5小时(pp)或3小时(pe)。
[0265]
通过ftir测定聚合物共聚单体(1-丁烯)含量
[0266]
使用bruker tensor 37光谱仪和opus软件基于傅里叶变换红外光谱(ftir)确定共聚单体含量。
[0267]
将约0.3克的样品压缩成型为厚度为250μm的薄膜。薄膜的两面均使用硅胶纸。为了避免污染,不徒手接触薄膜。使用fontijne press的型号labecon 300压片机来压制薄膜。成型在160℃下进行,预热2分钟+轻压2分钟+全压1分钟。冷却通过全压4分钟完成。
[0268]
丁烯共聚单体含量由约1378cm-1
波数处的吸光度来确定,参考峰为2019cm-1
。使用2cm-1
的分辨率、4000至400cm-1
的波数跨度和128的扫描次数进行分析。从每个薄膜中获得至少两个光谱。
[0269]
共聚单体含量由波数为1400cm-1
至1330cm-1
的光谱来确定。基线通过如下方法确定:在设定的波数范围内,定位最高峰,然后在该最高峰的左侧和右侧找到最小值。用基线连接这些最小值。用最高峰的吸光度值除以参考峰的面积。
[0270]
该方法的校准曲线针对每种共聚单体类型分别生成。未知样品的共聚单体含量需要在校准样品的共聚单体含量范围内。校准样品材料中的共聚单体含量通过nmr光谱法预先确定。
[0271]
使用校准曲线和下式自动计算共聚单体含量:
[0272]
we=c1×
a0+c0[0273]
式中
[0274]
we=结果(单位为wt%)
[0275]
a0=测量峰的吸光度(aq)/参考峰的面积(ar);
[0276]
c1=校准曲线的斜率;
[0277]
c0=校准曲线的偏移量。
[0278]
共聚单体含量由所获得的两个光谱确定,该值被计算为这些结果的平均值。
[0279]
实施例
[0280]
内部给体比较例
[0281]
cd1:二(呋喃-2-基)甲烷(dfm)
[0282]
[0283]
二(呋喃-2-基)甲烷(dfm)(cas:1197-40-6),也称为双(2-呋喃基)甲烷,其根据[a)chem.eur.j.,2000,6,22,4091;b)j.appl.polym.sci.,2014,doi:10.1002/app.40179]中所述地制备。产物通过真空蒸馏(沸点70℃/15毫巴)来分离,收率为62%。
[0284]1h nmr(cdcl3,400mhz):δ7.33(m,2h),6.31(m,2h),6.08(d,j=3.0hz,2h),4.00(s,2h)。
[0285]
13
c{1h}nmr(cdcl3,100mhz):δ151.5、141.6、110.4、106.4、27.4。
[0286]
cd2:1,1-二(呋喃-2-基)乙烷(dfe)
[0287][0288]
1,1-二(呋喃-2-基)乙烷(cas:51300-81-3),也称为1,1-双(2-呋喃基)乙烷或2,2'-(乙烷-1,1-二基)二呋喃,根据稍微修改的[j.heterocyclic.chem.,1991,28,991]中描述的方法制备。
[0289]
向冰冷却的85ml乙醇和50ml 12m hcl的混合物中加入175g(2.57mol)呋喃,然后加入60g(1.36mol)乙醛。将所得混合物在室温下搅拌20小时,然后倒入500ml水中,用3
×
250ml乙醚萃取。合并有机萃取物,用碳酸氢钾水溶液洗涤,用硫酸钠干燥,真空蒸发挥发物。真空蒸馏残余物,得到60.5g(29%)1,1-二(呋喃-2-基)乙烷(沸点88至92℃/17mm hg)和57.2g(26%)2,5-双[1-(2-呋喃基)乙基]呋喃(沸点144至152℃/3mm hg)。
[0290]1h nmr(cdcl3,400mhz):δ7.32(m,2h),6.29(dd,j=3.1hz,j=1.9hz,2h),6.04(d,j=3.1hz,2h),4.21(q,j=7.2hz,1h),1.59(d,j=7.2hz,3h)。
[0291]
13
c{1h}nmr(cdcl3,100mhz):δ156.5、141.3、110.1、104.9、33.0、17.9。
[0292]
cd3:2,2-二(呋喃-2-基)丙烷(dfp)
[0293][0294]
2,2-二(呋喃-2-基)丙烷(cas:17920-88-6),也称为2,2-双(2-呋喃基)丙烷或2,2'-(丙烷-2,2-二基)二呋喃,购自tci europe n.v.。
[0295]
cd4:三(呋喃-2-基)甲烷(tfm)
[0296]
[0297]
向冰冷却的8.6ml糠醛(103.8mmol)和200ml呋喃的混合物中滴加5ml三氟乙酸,所得混合物在室温下搅拌24小时。将反应混合物倒入碳酸氢钾水溶液中,用250ml乙醚萃取,萃取液用na2so4干燥。真空除去所有挥发物,真空蒸馏残余物,得到3.2g(14%)三(呋喃-2-基)甲烷(cas:77616-90-1),也称为2,2',2
”‑
甲烷三基三呋喃(沸点115℃/10mm hg)。
[0298]1h nmr(cdcl3,400mhz):δ7.40(br.d,3h),6.36(dd,j=3.1hz,j=1.9hz,3h),6.16(d,j=3.1hz,3h),5.59(s,1h)。
[0299]
13
c{1h}nmr(cdcl3,100mhz):δ151.9、142.0、110.4、107.3、38.9。
[0300]
cd5:2,5-双[2-(四氢呋喃-2-基)丙-2-基]四氢呋喃(bthfpt)
[0301][0302]
2,5-双[2-(四氢呋喃-2-基)丙烷-2-基]四氢呋喃(cas:89686-70-4),也称为2,5-双[2-(茂烷-2-基)丙烷-2-基]氧杂环戊烷,购自fluorochem有限公司。
[0303]
cd6:2,5-双[1-(呋喃-2-基)乙烷-1-基]呋喃(bfef)
[0304][0305]
2,5-双[1-(呋喃-2-基)乙烷-1-基]呋喃(cas:61093-50-3)使用稍微修改的[j.heterocyclic.chem.,1991,28,991]所述的方法制备。
[0306]
向冰冷却的85ml乙醇和50ml 12m hcl的混合物中加入175g(2.57mol)呋喃,然后加入60g(1.36mol)乙醛。将所得混合物在室温搅拌20小时,倒入500ml水中,用3
×
250ml乙醚萃取。合并有机萃取物,用碳酸氢钾水溶液洗涤,用硫酸钠干燥,真空蒸发挥发物。真空蒸馏残余物,得到60.5g(29%)2,2'-乙烷-1,1-2基二呋喃(沸点88至92℃/17mm hg)和57.2g(26%)2,5-双[1-(呋喃-2-基)乙烷-1-基]呋喃(沸点144至152℃/3mm hg)。
[0307]1h nmr(cdcl3,400mhz):δ7.31(m,2h),6.27(dd,j=3.1hz,j=1.9hz,2h),6.00(d,j=3.1hz,2h),5.93(s,2h),4.16(q,j=7.2hz,2h),1.56(d,j=7.2hz,6h)。
[0308]
13
c{1h}nmr(cdcl3,100mhz):δ156.7、155.2、141.1、110.0、105.4、104.9、33.1、18.01、17.96。
[0309]
cd7:2,2,7,7,12,12,17,17-八甲基-21,22,23,24-四氧杂过氢四烯(omtopq)
[0310][0311]
2,2,7,7,12,12,17,17-八甲基-21,22,23,24-四恶四烯
[0312]
依次向在200ml无水乙醇中的95.2g(0.4mol)cocl2(h2o)6溶液中加入93.2g(1.6mol)丙酮、32ml 12m hcl,最后加入54.4g(0.8mol)呋喃。约20分钟后,反应混合物变热(60至70℃)并变成深红色。将反应烧瓶浸入水浴中,在60℃下保持3小时。然后将反应混合物在室温下搅拌过夜,倒入500ml水中。所得悬浮液用3
×
150ml甲苯萃取。对合并萃取物的挥发物进行真空蒸发,将残余物与200ml无水乙醇一起研磨,得到11.1g(13%,纯度》95%)所需产物,为白色粉末。粗产物从热甲苯中重结晶,得到分析纯的2,2,7,7,12,12,17,17-八甲基-21,22,23,24-四恶四烯,为白色固体。
[0313]1h nmr(cdcl3,400mhz):δ5.88(s,1h),1.47(s,3h)。
[0314]
2,2,7,7,12,12,17,17-八甲基-21,22,23,24-四氧杂过氢四烯
[0315]
将2,2,7,7,12,12,17,17-八甲基-21,22,23,24-四恶四烯(5.0g,11.6mmol)、5%pd/c(710g)和乙醇(170ml)置于1000ml高压釜中,然后用氢气(100巴)对其加压。将该混合物在125℃搅拌15小时。冷却至室温后,形成的混合物通过celite 503垫用额外100ml乙醇洗涤过滤。将滤液挥发物真空蒸发至干,得到第一部分的所需产物(2.25g),其为11种非对映异构体的混合物(gc-ms)。滤饼用400ml二氯甲烷洗涤,滤液挥发物在真空中蒸发至干,得到第二部分的产物(2.90g),其为两种主要非对映异构体与少量两种次要非对映异构体的混合物(gc-ms)。将产物的两部分合并。总收率为5.15g(99%)的2,2,7,7,12,12,17,17-八甲基-21,22,23,24-四氧杂过氢四烯(cas:50451-63-3),也称为八甲基全氢环四呋喃,为白色固体。
[0316]
合并部分的1h nmr(cdcl3,400mhz):δ4.20-3.20(m,8h),2.55-1.28(m,16h),1.26-0.60(m,24h)。
[0317]
cd8:9,9-二(甲氧基甲基)芴(dmmf)
[0318][0319]
9,9-二(甲氧基甲基)芴(cas:182121-12-6),也称为9,9-双(甲氧基甲基)-9h-芴,
购自杭州赛捷化工有限公司。
[0320]
内部给体发明例
[0321]
给体发明例id1至id4根据如下步骤制备:
[0322]
id1:二(四氢呋喃-2-基)甲烷(dthfm)
[0323][0324]
将2,2'-亚甲基二呋喃(19.2g,130mmol;参见cd1的制备)和5%pd/c(0.95g)放入用氩气吹扫的450ml高压釜中,用氢气(70巴)加压,将混合物在100℃搅拌1小时。冷却至室温并减压后,混合物用二氯甲烷稀释,通过celite 503垫过滤。滤液挥发物在真空中蒸发。之后,将乙酸(20ml)和水(80ml)加入到残余物中,将形成的混合物在氩气氛下回流2小时。冷却至室温后,用稍过量的naoh碱化,用己烷萃取由此得到的混合物。萃取液用na2so4干燥,真空蒸发溶剂。在氩气氛下将残余物在搅拌下滴加到2.4g nah中。当氢气停止形成时,真空蒸馏产物(沸点54至70℃/1毫巴)。收率:11.4g(56%)无色液体。根据1h nmr,产物是二(四氢呋喃-2-基)甲烷(cas:1793-97-1)的非对映异构体a和b的约1:1混合物,也称为2,2'-亚甲基双(四氢呋喃),其中a是(l)和(d)异构体的外消旋混合物,b是内消旋异构体。
[0325]1h nmr(cdcl3,400mhz):δ3.95-3.79(m,a中4h以及b中4h),3.72-3.65(m,a中2h以及b中2h),2.05-1.93(m,a中2h以及b中2h),1.92-1.76(m,a中4h以及b中5h),1.69(t,j=6.4hz,a中2h),1.59(dt,j=13.6hz,j=5.8hz,b中1h),1.52-1.39(m,a中2h以及b中2h)。
[0326]
13
c{1h}nmr(cdcl3,100mhz):δ77.1、76.9、67.7、67.5、41.7、41.1、31.9、31.6、25.6、25.5。
[0327]
id2:1,1-二(四氢呋喃-2-基)乙烷(dthfe)
[0328][0329]
将2,2'-(乙烷-1,1-二基)二呋喃(20g,123mmol;参见cd2)和5%pd/c(1.00g)的混合物放入450ml高压釜中,然后用氩气吹扫、用氢气加压(70巴)。将该混合物在120℃搅拌1小时,冷却至室温并减压后,用二氯甲烷稀释混合物,通过celite 503垫过滤。滤液挥发物在真空中蒸发。然后,往残余物中加入乙酸(20ml)和水(80ml),混合物在氩气氛下回流5小时。冷却至室温后,用稍过量的naoh碱化,用己烷萃取。萃取液用na2so4干燥,真空蒸发溶剂。在氩气氛下将残余物在搅拌下滴加到3g nah中。当氢气停止形成时,真空蒸馏产物(沸点75至90℃/4毫巴)。收率:8.4g(40%)1,1-二(四氢呋喃-2-基)乙烷(cas:84548-20-9),也称为2,2'-亚乙基双(四氢呋喃),为无色液体。根据1h nmr,产物是约2.2:1.1:1的非对映异构体a、b和c的混合物。
[0330]1h nmr(cdcl3,400mhz):δ3.96-3.90(m,b中2h),3.86-3.76(m,a中6h,b中4h,c中6h),1.98-1.44(m,a,b和c中9h),0.97(d,j=6.9hz,b中3h),0.87(d,j=6.8hz,a中3h),
0.80(d,j=6.9hz,c中3h)。
[0331]
13
c{1h}nmr(cdcl3,100mhz):δ81.4、81.1、81.0、80.1、68.0、67.9、67.8、67.7、42.4、42.0、41.3、29.4、29.3(两种共振态)、27.9、26.1、25.9、25.7(两种共振态)、10.6、10.3、10.2。
[0332]
id3和id4:分别为内消旋-三(四氢呋喃-2-基)甲烷(tthfm-a)和外消旋-三(四氢呋喃-2-基)甲烷(tthfm-b)
[0333][0334]
三(呋喃-2-基)甲醇
[0335]
在-10℃下,向呋喃(87.3g,1.28mol)的thf(1000ml)溶液中滴加正丁基锂(500ml,1.25mol,2.5m溶于己烷)。将反应混合物升温至10℃,在该温度搅拌4小时。之后,将形成的2-呋喃基锂悬浮液冷却至-80℃,加入碳酸二甲酯(28.8g,320mmol)。将混合物在室温搅拌过夜,倒入1l水中,用3
×
300ml乙醚萃取。合并的萃取物用200ml水洗涤,经na2so4干燥,通过硅胶60垫(40至63μm),在真空中蒸发挥发物至干。该程序获得62.0g 90%纯度的三(呋喃-2-基)甲醇(76%收率),为深棕色油状物,其无需进一步纯化即可用于下一阶段。
[0336]1h nmr(cdcl3,400mhz):δ7.43(d,3h,j=1.8hz),6.38-6.36(dd,3h,j=3.2hz,j=1.7hz),6.23(d,3h,j=3.2hz),3.38(s,1h)。
[0337]
2,2'-(呋喃-2(5h)-亚基亚甲基)二呋喃
[0338]
在0℃下向三氯化铝(23.2g,174mmol)在无水乙醚(1000ml)中的溶液中分批加入氢化铝锂(16.0g,416mmol)。向得到的溶液中滴加三(呋喃-2-基)甲醇(40.0g,174mmol)。将混合物在0℃搅拌2小时,用100ml水小心淬灭。获得的悬浮液通过celite 503垫过滤,滤液用na2so4干燥,挥发物在真空中蒸发至干。对残余物进行分馏,得到26.1g(70%)2,2'-(呋喃-2(5h)-亚基亚甲基)二呋喃,为浅棕色油状物(沸点145℃/5毫巴),根据1h nmr,其含有约5mol%2,2',2
”‑
甲烷三基三呋喃。
[0339]1h nmr(cdcl3,400mhz):7.43(m,1h),7.38(m,1h),6.70(dt,1h,j=6.1hz,j=2.3hz),6.63(d,1h,j=3.3hz)),6.49(dt,1h,j=6.1hz,j=2.3hz),6.44(m,2h),6.36(d,1h,j=3.1hz),5.20(m,2h)。
[0340]
内消旋-三(四氢呋喃-2-基)甲烷(tthfm-a)和外消旋-三(四氢呋喃-2-基)甲烷(tthfm-b)
[0341]
将2,2'-(呋喃-2(5h)-亚基亚甲基)二呋喃(3.6g,16.8mmol)、5%pd/c(180mg)和4ml i
proh的混合物置于100ml用氩气吹扫的高压釜中,用氢气加压(70巴),混合物在100℃搅拌75分钟。冷却至室温并减压后,将形成的混合物用二氯甲烷稀释,通过celite 503垫过滤。真空蒸发滤液的挥发物,通过硅胶60(40至63μm,洗脱液:己烷/乙酸乙酯,3:1,体积)柱色谱纯化残余物。该程序提供了分离的立体异构体形式的三(四氢呋喃-2-基)甲烷:0.77g
内消旋非对映异构体tthfm-a和0.28g外消旋非对映异构体tthfm-b。两种非对映异构体的总收率为1.05g(28%)。除此之外,分离出0.78g(22%)的3-[二(四氢呋喃-2-基)甲基]呋喃。
[0342]
非对映异构体tthfm-a。1h nmr(cdcl3,400mhz):δ4.06-3.96(m,2h),3.92-3.86(m,1h),3.86-3.77(m,3h),3.70-3.63(m,3h),2.10-2.05(m,1h),2.01-1.66(m,12h)。
[0343]
13
c{1h}nmr(cdcl3,100mhz):δ78.1、78.0、77.9、67.6、67.4(两种共振态)、49.2、30.4、29.9、29.0、26.0、25.8(两种共振态)。
[0344]
非对映异构体tthfm-b。1h nmr(cdcl3,400mhz):δ3.98-3.94(m,3h),3.86-3.79(m,3h),3.69-3.62(m,3h),2.00-1.62(13h)。
[0345]
13
c{1h}nmr(cdcl3,100mhz):δ78.6、62.7、50.5、30.6、25.8。
[0346]
催化剂组分发明例和比较例
[0347]
根据wo 2016/124676 a1的参考例2的程序制备催化剂组分发明例(ic1-ic4)和催化剂组分比较例(cc1-cc8),但分别使用给体发明例(id1-id4)和给体比较例(cd1-cd8)。cc8-a和cc8-b中使用相同的内部给体cd8。表1中公开了催化剂组分实施例中所用给体的概述。
[0348]
催化剂组分比较例cc9是以商品名lynx 200购自grace的催化剂。
[0349]
原材料
[0350]
通过对购自chemtura的100wt%tea-s进行稀释来制备溶于庚烷的10wt%tea(三乙基铝)储备溶液。
[0351]
mgcl2*3etoh载体购自grace。
[0352]
ticl4购自aldrich(金属杂质《1000ppm,金属分析》99.9%)。
[0353]
催化剂组分制备的一般程序
[0354]
在惰性气氛手套箱中,向干燥的100ml四颈圆底烧瓶(配备有两个橡胶隔片、一个温度计和一个机械搅拌器)中装入溶解在40ml庚烷中的3.1mmol的所需内部给体和7.01g(30mmol的mg)的颗粒状(d
v0.5
为17μm)mgcl2*2.93etoh载体(内部给体和内部给体/mg加样比如表1和表2中所示)。从手套箱中取出烧瓶,连接氮气入口和出口。将烧瓶置于冷却浴中,在0℃以250rpm缓和约10分钟。在1小时内将三乙基铝在庚烷(107.55g,94.2mmol al;al/etoh=1.07mol/mol)中的10wt%溶液滴加到搅拌的悬浮液中,保持反应混合物温度低于0℃。将获得的悬浮液在20分钟内加热至80℃,在该温度下以250rpm的速度再保持30分钟。将悬浮液在80℃下静置5分钟,使用套管去除上清液。在室温下,将获得的预处理载体材料用70ml甲苯洗涤两次(加入甲苯,以250rpm搅拌15至120分钟,沉降5分钟,虹吸掉液相)。
[0355]
在室温下,将70ml甲苯加入预处理的载体材料中。向以250rpm搅拌的悬浮液中逐滴加入纯ticl4(3.31ml,30mmol;ti/mg=1.0mol/mol),反应混合物温度被保持为25至35℃。将获得的悬浮液在20分钟内加热至90℃,在该温度下以250rpm的速度再搅拌60分钟。将悬浮液在90℃下静置5分钟,使用套管去除上清液。将得到的催化剂在90℃下用70ml甲苯洗涤两次,在室温下用70ml庚烷洗涤一次(每次洗涤包括加入甲苯或庚烷,以250rpm搅拌15分钟,沉降5分钟,虹吸掉液相)。将催化剂在70℃下真空干燥30分钟。
[0356]
表1:催化剂组分cc1至cc8和ic1至ic4中使用的给体概述
[0357]
实施例催化剂组分内部给体内部给体缩写
比较例cc1cd1dfm比较例cc2cd2dfe比较例cc3cd3dfp比较例cc4cd4tfm比较例cc5cd5bthfpt比较例cc6cd6bfef比较例cc7cd7omtopq比较例cc8-acd8dmmf比较例cc8-bcd8dmmf发明例ic1id1dthfm发明例ic2id2dthfe发明例ic3id3tthfm-a发明例ic4id4tthfm-b
[0358]
比较例
[0359]
cc1制备
[0360]
使用上述一般程序来制备cc1,不同之处在于使用0.45g(3.1mmol)的cd1(dfm)作为内部给体(id/mg=0.102)。
[0361]
分离出3.8g(94.8%收率,镁系)的cc1。
[0362]
cc2制备
[0363]
使用上述一般程序来制备cc2,不同之处在于使用0.50g(3.1mmol)的cd2(dfe)作为内部给体(id/mg=0.102)。
[0364]
分离出3.6g(82.4%收率,镁系)的cc2。
[0365]
cc3制备
[0366]
使用上述一般程序来制备cc3,不同之处在于使用0.54g(3.1mmol)的cd3(dfp)作为内部给体(id/mg=0.102)。
[0367]
分离出3.0g(68.3%收率,镁系)的cc3。
[0368]
cc4制备
[0369]
使用上述一般程序来制备cc4,不同之处在于使用0.66g(3.1mmol)的cd4(tfm)作为内部给体(id/mg=0.102)。
[0370]
分离出3.8g(78.2%收率,镁系)的cc4。
[0371]
cc5制备
[0372]
使用上述一般程序来制备cc5,不同之处在于使用0.34g(1.2mmol)的cd5(bthfpt)作为内部给体(id/mg=0.039)。
[0373]
分离出3.3g(85.5%收率,镁系)的cc5。
[0374]
cc6制备
[0375]
使用上述一般程序来制备cc6,不同之处在于使用0.78g(3.1mmol)的cd6(bfef)作为内部给体(id/mg=0.102)。
[0376]
分离出4.0g(89.9%收率,镁系)的cc6。
[0377]
cc7制备
[0378]
使用上述一般程序来制备cc7,不同之处在于使用1.37g(3.1mmol)的cd7(omtopq)作为内部给体(id/mg=0.102)。
[0379]
分离出5.9g(95.5%收率,镁系)的cc7。
[0380]
cc8-a制备
[0381]
使用上述一般程序来制备cc8-a,不同之处在于使用0.778g(3.1mmol)的cd8(dmmf)作为内部给体(id/mg=0.102)。
[0382]
分离出2.3g(57.4%收率,镁系)的cc8-a。
[0383]
cc8-b制备
[0384]
使用上述一般程序来制备cc8-b,不同之处在于使用1.038g(4.1mmol)的cd8(dmmf)作为内部给体(id/mg=0.136)。
[0385]
分离出2.8g(64.5%收率,镁系)的cc8-b。
[0386]
发明例
[0387]
ic1制备
[0388]
使用上述一般程序来制备ic1,不同之处在于使用0.48g(3.1mmol)的id1(dthfm)作为内部给体(id/mg=0.102)。
[0389]
分离出3.9g(84.0%收率,镁系)的ic1。
[0390]
ic2制备
[0391]
使用上述一般程序来制备ic2,不同之处在于使用0.52g(3.1mmol)的id2(dthfe)作为内部给体(id/mg=0.102)。
[0392]
分离出4.7g(93.4%收率,镁系)的ic2。
[0393]
ic3制备
[0394]
使用上述一般程序来制备ic3,不同之处在于使用0.69g(3.1mmol)的id3(tthfm-a)作为内部给体(id/mg=0.102)。
[0395]
分离出5.2g(96.9%收率,镁系)的ic3。
[0396]
ic4制备
[0397]
使用上述一般程序来制备ic4,不同之处在于使用0.46g(2.0mmol)的id4(tthfm-b)作为内部给体(id/mg=0.068)。
[0398]
分离出4.7g(96.7%收率,镁系)的ic4。
[0399]
表2:催化剂组分cc1至cc8和ic1至ic4的性质概述
[0400][0401]
与1-丁烯的共聚(实验室规模)
[0402]
测试了催化剂组分发明例(ic1-ic4)和催化剂组分比较例(cc1至cc7、cc8-a、cc8-b和cc9)与1-丁烯的共聚(ie1至ie4、ce1至ce7、ce8-a、ce8-b和ce9-a至ce9-c)。使用三乙基铝(tea)作为助催化剂,其中al/ti摩尔比为15。聚合反应在3l实验室规模反应器中按照如下程序进行:
[0403]
在20℃下,将70ml的1-丁烯装入空的3l实验室规模反应器,以200rpm搅拌。然后将1250ml丙烷加入反应器中作为聚合介质,然后添加氢气(0.40巴)。将反应器加热至85℃,分批加入乙烯(3.7巴)。反应器压力保持在0.2巴的超压,搅拌速度增加到550rpm。将催化剂组分和助催化剂一起加入反应器中(催化剂组分与tea预接触几秒钟)并加有额外的100ml丙烷。通过连续的乙烯进料将总反应器压力保持在37.8巴。60分钟后通过排出单体和h2以停止聚合。将得到的聚合物放置在通风橱中干燥过夜,然后称重。
[0404]
此外,还测试了催化剂组分比较例cc9与55ml 1-丁烯和0.40巴氢气(ce9-b)的共聚测试以及与40ml 1-丁烯和0.75巴氢气(ce9-c)的共聚测试。
[0405]
聚合结果
[0406]
聚合反应的结果示于表3。催化剂的活性根据催化剂组分加样量和一小时内产生的聚合物的量来计算。
[0407]
表3:比较例ce1至ce7、ce8-a至ce8-b、ce9-a至ce9-c和发明例ie1至ie4的聚合结果概述
[0408][0409]
由表3的结果和图1至图3所示,所有发明例(ie1至ie4)与比较例ce1至ce9(a至c)相比具有更窄的mwd和更高的mw。
[0410]
此外,所有发明例ie1至ie4均显示出与比较例ce1至ce9(a-c)相比更低的给定共聚单体含量下的熔融温度。
[0411]
所有发明例ie1至ie4均具有与比较例ce1至ce8相似的活性水平,但具有高得多的mw能力。
[0412]
此外,所有发明例ie1至ie4均实现了所需的性能平衡,其中发明例的id/mg加样比与内部给体比较例cd8必须采用的id/mg加样比相似或更低。
[0413]
综上发现,所有发明例ie1至ie4均显示出最高的mw与非常窄的mwd和给定共聚单体含量下的最低的熔融温度的组合(在催化剂组分/内部给体的测试组中)。因此,本发明的内部给体允许定制所生产聚合物的性质,同时将催化剂生产率保持在可接受的高水平。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1