利用调控Dahms途径转化木糖生成fengycin的合成菌株构建方法
regulation strategies of nrpss for biosynthesis of lipopeptides by bacillus[j].applied microbiology and biotechnology,2020,104(19):8077
‑
8087)。但是由于fengycin的生物合成底物经济适用性不足等因素,关于fengycin在其它的方面的应用和功能的探究受到了很大的限制(kumar pn,swapna th,khan my,et al.statistical optimization of antifungal iturin a production from bacillus amyloliquefaciens rhnk22 using agro
‑
industrial wastes[j].saudi journal of biological sciences,2015,370(s 5
‑
6):636
–
640.),因此有必要构建通过廉价碳源转化fengycin的菌株,而木糖作为木质纤维素中的第二大单糖,是作为碳源最好的选择。
[0004]
fengycin合成的前体中包含多种的氨基酸和脂肪酸,在初级的碳代谢过程中,丙酮酸是连接氨基酸代谢、脂肪酸延伸以及中心碳代谢的主要节点代谢物,因此提高从碳源到丙酮酸的转化速率,有利于fengycin的相关的前体的合成。在目前已经发现的自然界中的木糖的代谢途径中,传统的异构化和氧化还原途径转化木糖到丙酮酸都需要首先经过磷酸戊糖途径和糖酵解途径,最近发现的非磷酸化木糖途径weimberg直接转化木糖到α
‑
酮戊二酸,不经过丙酮酸,而另一代谢途径dahms,仅4步反应就可以完成从木糖到丙酮酸的过程(valdehuesa kng,ramos krm,nisola gm,et al.everyone loves an underdog:metabolic engineering of the xylose oxidative pathway in recombinant microorganisms[j].applied microbiology and biotechnology,2018,102(18):7703
‑
7716.),因此选择dahms木糖代谢途径作为fengycin合成的碳源摄取通道具有一定的探究意义。但是在利用dahms途径转化木糖的研究中发现,该木糖代谢途径的表达会使菌株的生物量降低,比如so young choi等在e.coli xl1
‑
blue中引入来自c.crescentus的基因xylb和xylc成功构建了完整的dahms代谢途径,在发酵过程中观察到相较于原始菌株,菌株的生长状况变差,而且产生了一定量的乳酸积累(choi sy,park sj,kim wj,et al.one
‑
step fermentative production of poly(lactate
‑
co
‑
glycolate)from carbohydrates in escherichia coli[j].nature biotechnology,2016,34(4):435
‑
+.)。同时,dahms途径还会生成毒性代谢物乙醇醛,给细胞带来毒性作用。因而,在引入dahms途径转化木糖的同时,还需要考虑dahms途径的表达带来的负面影响,在消除乙醇醛的毒性作用的同时将其转化为tca循环中的中间物质,加强tca循环通量的同时间接改善菌株的胞内代谢状态。
[0005]
本发明综合利用合成生物学、基因工程和代谢工程技术,对涉及dahms木糖代谢途径设计模块制备,并设计将dahms途径表达副产物乙醇醛出发的调控基因进行重组,从而产生优质的稳定利用木糖合成fengycin的合成菌株。
技术实现要素:
[0006]
本发明的目的是通过合成生物学等的技术手段从头设计制备了木糖的dahms途径模块,并以dahms途径的表达毒性产物乙醇醛出发设计了针对dahms模块的调控乙醇醛回补tca模块,构建以dahms模块和调控乙醇醛回补tca模块同时组合表达转化木糖合成fengycin的优质合成菌株bsu02,利用木糖为碳源发酵,检测fengycin的产量为46.82mg/l。
[0007]
本发明的目的是通过以下的技术方案实现:
[0008]
一种利用调控dahms途径转化木糖生成fengycin的合成菌株构建方法;以dahms途径模块与调控乙醇醛回补tca模块组合表达转化木糖合成fengycin的优质菌株的构建方
法;从fengycin的合成前体供应出发,将来自枯草芽孢杆菌的启动子基因p
43
和来自新月柄杆菌的木糖脱氢酶xylb、木糖酸内酯酶xylc以及来自大肠杆菌mg1655的木糖酸脱水酶基因yjhg、2
‑
酮
‑3‑
脱氧木糖酸脱水酶基因yjhh串联,制备dahms木糖途径模块到穿梭载体phy300plk中;同时从dahms途径的代谢物乙醇醛出发,将来自大肠杆菌得醛脱氢酶基因alda、苹果酸合成酶aceb和苹果酸脱氢酶mdh和启动子p
43
串联,制备调控乙醇醛回补tca调控模块到穿梭载体php13载体中;将两个穿梭载体在底盘菌株中共表达,构建以dahms模块和调控乙醇醛回补tca模块共表达转化木糖合成fengycin的优质菌株bsu02。
[0009]
本发明所述的方法包括步骤如下:
[0010]
1)根据p
43
启动子设计pcr引物,以bacillus subtilis 168基因组为模板,扩增基因片段启动子p
43
;
[0011]
2)将启动子p
43
连入穿梭载体phy300plk,获得重组载体phy300p01;
[0012]
3)根据xylb基因设计pcr引物,扩增木糖脱氢酶基因xylb;
[0013]
4)根据xylc基因设计pcr引物,,扩增木糖酸内酯酶基因xylc;
[0014]
5)将扩增基因xylb和扩增基因xylc通过无缝克隆连接体系连入步骤2)中构好的载体phy300p01,获得重组载体phy300p02;
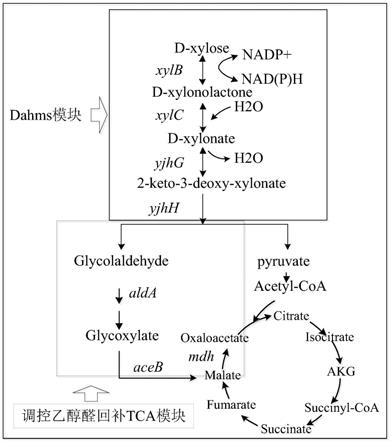
[0015]
6)根据yjhg基因设计pcr引物,以escherichia coli k
‑
12 mg1655基因组为模板,扩增木糖酸脱水酶基因yjhg;
[0016]
7)根据yjhh基因设计pcr引物,以escherichia coli k
‑
12 mg1655基因组为模板,扩增2
‑
酮
‑3‑
脱氧木糖酸脱水酶基因yjhh;
[0017]
8)将扩增基因yjhg和扩增基因yjhh通过无缝克隆体系连入步骤5)构好的载体phy300p02,得到重组载体phy300p03;
[0018]
9)根据p
43
启动子设计pcr引物,以bacillus subtilis 168基因组为模板,扩增基因片段启动子p
43
;
[0019]
10)将扩增启动子p
43
连入穿梭载体php13,获得重组载体php13p01;
[0020]
11)根据aceb基因设计pcr引物,以escherichia coli k
‑
12 mg1655基因组为模板,扩增苹果酸合成酶基因aceb;
[0021]
12)根据mdh基因设计pcr引物,以escherichia coli k
‑
12 mg1655基因组为模板,扩增苹果酸脱氢酶基因mdh;
[0022]
13)将扩增基因aceb和扩增基因mdh通过无缝克隆体系连入步骤10)构好的载体php13p01,得到重组载体php13p02;
[0023]
14)根据alda基因设计pcr引物,以escherichia coli k
‑
12mg1655基因组为模板,扩增醛脱氢酶基因alda;
[0024]
15)扩增基因alda通过无缝克隆体系连入步骤13)构好的载体php13p02,得到重组载体php13p03;
[0025]
16)将步骤8)构好的重组载体phy300p03和步骤15)构好的重组载体php13p03通过电转化的方法导入b.subtilis 168改造菌中,构建dahms模块和调控乙醇醛回补tca模块组合表达转化木糖合成fengycin的优质菌株bus02。
[0026]
所述的p
43
启动子的已知序列seq id no:1。
[0027]
所述的xylb基因的已知序列seq id no:2。
mg1655基因组为模板,扩增苹果酸脱氢酶基因mdh;
[0048]
13)通过无缝克隆体系将扩增基因aceb和扩增基因mdh连入内切酶xhoi
‑
ecori双酶切处理后的线性载体php13p01,构建重组载体php13p02;
[0049]
14)alda是escherichia coli k
‑
12 mg1655菌中编码醛脱氢酶的基因,其核苷酸序列如seq id no:8所示,根据此序列设计pcr扩增引物,以escherichia coli k
‑
12 mg1655基因组为模板,扩增醛脱氢酶基因alda;
[0050]
15)通过无缝克隆体系将基因alda连入经过sphi
‑
xhoi双酶切处理的载体php13p02,构建重组载体php13p03;
[0051]
16)通过电转化的方式将步骤8)构建好的重组载体phy300p03和步骤15)构好的重组载体php13p03导入到b.subtilis 168改造菌中,构建以dahms模块和调控乙醇醛模块组合表达转化木糖合成fengycin的优质菌株bsu02;
[0052]
17)利用木糖生产fengycin,将16)构好的菌株用于摇瓶发酵培养。
[0053]
利用本发明提出的方法构建的工程菌株bsu02能够有效的利用木糖合成fengycin,高效液相色谱检测bsu02发酵液中的fengycin产量达到46.82mg/l,本发明提出的工程菌株的构建方法为以木糖为底物合成fengycin产量提供了一种新的策略。
附图说明
[0054]
图1:质粒phy300p01的酶切和pcr验证
[0055]
图2:xylb、xylc的pcr扩增和质粒phy300p02的酶切验证
[0056]
图3:质粒phy300p03的验证
[0057]
图4:质粒php13p01的验证
[0058]
图5:质粒php1302的验证
[0059]
图6:质粒php1303的验证
[0060]
图7:dahms模块载体phy300p03的构建流程
[0061]
图8:调控乙醇醛回补tca模块质粒载体php13p03的构建流程
[0062]
图9:dahms模块和调控乙醇醛回补tca模块的代谢图
[0063]
具体的实施方式
[0064]
一种以dahms途径模块与调控乙醇醛回补tca模块组合表达转化木糖合成fengycin的优质菌株的构建方法;其特征在于从fengycin的合成前体供应出发,为了缩短碳源木糖到节点代谢物丙酮酸的代谢步骤,利用基因工程的技术,将来自枯草芽孢杆菌的启动子基因p
43
和来自新月柄杆菌的木糖脱氢酶xylb、木糖酸内酯酶xylc以及来自大肠杆菌mg1655的木糖酸脱水酶基因yjhg、2
‑
酮
‑3‑
脱氧木糖酸脱水酶基因yjhh串联,制备dahms木糖途径模块到穿梭载体phy300plk中,同时为了将dahms途径的毒性代谢物乙醇醛进行有效再利用,将来自大肠杆菌得醛脱氢酶基因alda、苹果酸合成酶aceb和苹果酸脱氢酶mdh和启动子p
43
串联,制备调控乙醇醛回补tca调控模块到穿梭载体php13载体中。将两个穿梭载体在底盘菌中共表达,构建以dahms模块和调控乙醇醛回补tca模块共表达转化木糖合成fengycin的优质菌株bsu02,利用木糖为碳源发酵,检测fengycin的产量46.82mg/l。
[0065]
实施例一:dahms模块表达载体phy300p03的构建
[0066]
(1)启动子p
43
(seq id no:1)和基因xylb(seq id no:2)、xylc(seq id no:3)、
yjhg(seq id no:4)、yjhh(seq id no:5)基因扩增引物的设计及合成。在穿梭载体phy300plk中可用多克隆位点比较少,因此在设计p
43
的引物时,同时在引物的3端添加sphi、xhoi内切酶识别序列。根据基因xylb、xylc、yjhg、yjhh的核苷酸序列设计含rbs序列的扩增引物如下(下划线表示hindiii酶切位点,双下划波浪线表示xhoi酶切位点,单波浪线表示sphi,虚线表示sali,双下划线表示rbs序列),质粒引入设计如下,引物由擎科生物有限公司进行合成。
[0067]
p43
‑
f:aacggctttgcccaagctttgataggtggtatgttttcgcttga
[0068]
p43
‑
r:r:
[0069][0070]
xylb
‑
r:acgtaacttgcgcggtcatagctataccctccttggttagcgccagcccgcatcg
[0071]
xylc
‑
f:
[0072]
xylc
‑
r:ttataacaggaattcttaaaccagacgaacttcatgctgtggct
[0073]
yjhg
‑
f:
[0074]
yjhg
‑
r:aatattgcgaacagacatagctataccctccttggtcagactggtaaaatgcc
[0075]
yjhh
‑
f:
[0076]
yjhh
‑
r:aattgatcctttttttataacagcgagctctcagtttttattcataaaatcgcgc
[0077]
phy300
‑
f:agcggaatgacaccggtaaaccgaa
[0078]
phy300
‑
r:aggaatcattgtcattagttggctg
[0079]
(2)乙醇醛回补tca模块表达载体php13p03的构建。基因alda(seq is no:8)、aceb(seq is no:6)和mdh(seq is no:7)扩增引物设计及合成。根据alda、aceb和mdh基因的核苷酸序列,分别设计含rbs序列的扩增引物如下(下划线表示hindiii酶切位点,双下划波浪线表示xhoi酶切位点,单波浪线表示sphi,虚线表示sali,双下划线表示rbs序列),质粒php13的引物设计如下,引物由擎科生物科技有限公司进行合成。
[0080]
php13p43
‑
f:
[0081]
php13p43
‑
r:
[0082]
aceb
‑
f:
[0083]
aceb
‑
r:gaggactgcgactttcatagctataccctccttgggagctcttacgctaacaggcggt
[0084]
mdh
‑
f:
[0085]
mdh
‑
r:ttgtaaaacgacggccagtgaattcttacttattaacgaactctt
[0086]
alda
‑
f:
[0087]
alda
‑
r:agtcatagtagttcctccttatgtctcgagttaagactgtaaataaaccacctgg
[0088]
php13
‑
f:aagcggaagagcgcccaatacgca
[0089]
php13
‑
r:gtgatggttatcatgcaggattgt
[0090]
(3)escherichia coli mg1655(nc_000913.3)及bacillus subtilis 168(nc_000964.3)基因组的提取。将在lb液体培养基中220rpm,37℃过夜培养,获得新鲜的菌液用于基因组的提取,收集培养的细菌菌液大约1
‑
5ml,12000r/min离心并弃掉上清收集菌体;加1ml无菌水充分吹吸悬浮菌体,12000r/min离心30s,目的是为了防止培养基成分残留,该步骤重复2
‑
3次;向上一步得到的菌体中加入100ul破菌缓冲液吹吸重悬,并漩涡震荡3min;加入称量好的0.2g的石英砂,加入200ul的苯酚:氯仿:异丙醇=25:24:1的基因组提取液,再次漩涡震荡3min;吸取10ul的10
×
te缓冲液加入混匀,并漩涡震荡3min;12000r/min离心5min,小心吸取上清液到干净无菌的1.5ml离心管中;向得到的上清液添加20ul 3m的醋酸钠溶液和900ul的无水乙醇,轻轻上下颠倒混匀后,室温静置30min;12000r/min离心弃上清,并用70%的乙醇重悬洗涤沉淀3
‑
4次;12000r/min离心10min,收集沉淀,尽量倒净离心管内的液体,室温晾干至完全无酒精残留,加入20
‑
50ul的ddh2o溶解沉淀,在
‑
20℃条件下保存。
[0091]
(4)启动子p
43
、基因xylb、xylc、yjhg和yjhh、aceb、mdh、alda的扩增。以上述提取的bacillus subtilis 168的基因组为模板,以上述合成的p43
‑
f及p43
‑
r为引物扩增p
43
基因;以seq id no:2和seq id no:2序列为模板,利用引物xylb
‑
f/r、xylc
‑
f/r扩增xylb、xylc基因;以上述提取的escherichia coli mg1655基因组为模板,以上述合成的yjhg
‑
f/yjhg
‑
r、yjhh
‑
f/yjhh
‑
r、aceb
‑
f/aceb
‑
r、mdh
‑
f/mdh
‑
r、alda
‑
f/alda
‑
r为引物pcr扩增yjhg、yjhh、aceb、mdh、alda基因。
[0092]
pcr的反应体系为:pcr反应体系为:总体积50μl,无菌水17μl,2
×
phanta max buffer 25μl,dntp mix 1μl,上游引物2μl,下游引物2μl,phanta max super fidelity dna polymerase 1μl,模板2μl;pcr反应程序为:95℃3min,95℃15s,72℃60s/kb,72℃5min,4℃保温,循环数:30。
[0093]
(5)将启动子p
43
连入载体phy300plk。将载体进行hindiii和sali双酶切反应,用无缝克隆连接体系将p
43
启动子基因片段与线性化载体phy300plk连接。
[0094]
(6)载体phy300p01的筛选。将连接产物转入大肠杆菌dh5α感受态中,涂布于含100μg/ml的氨苄青霉素抗性的lb固体培养基(蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l,琼脂20g/l),37℃培养16
‑
18小时,挑取转化子进行菌落pcr验证,验证片段与理论片段相符合,即筛选出正确的重组载体phy300p01,如附图1所示,为载体hindiii
‑
xhoi双酶切和质粒引物phy300
‑
f/r的pcr扩增验证,电泳结果与理论长度4895bp、280bp以及644bp一致。
[0095]
(7)将基因xylb、xylc基因连入载体phy300p01。将载体进行xhoi和ecori内切酶反应,用无缝克隆连接体系将xylb和xylc连入线性化的质粒phy300p01。
[0096]
(8)载体phy300p02的筛选。将连接产物转化入大肠杆菌dh5α感受态中,涂布于含100μg/ml的氨苄青霉素抗性的lb固体培养基(蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l,琼脂20g/l),37℃培养16
‑
18小时,挑取转化子进行菌落pcr验证,验证片段与理论片段相符合,即筛选出正确的重组载体phy300p02,如附图2,为基因xylb、xylc的pcr扩增验证和载体phy300p02的hindiii
‑
ecori酶切验证,与理论序列长度相符。
[0097]
(9)将基因yjhg、yjhh基因连入载体phy300p02。将载体进行ecori单酶切反应,用无缝克隆连接体系将yjhg和yjhh连入线性化的质粒phy300p02。
[0098]
(10)载体phy300p03的筛选。将连接产物转化入大肠杆菌dh5α感受态中,涂布于含100μg/ml的氨苄青霉素抗性的lb固体培养基(蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l,琼脂20g/l),37℃培养16
‑
18小时,挑取转化子进行菌落pcr验证,验证片段与理论片段相符合,即筛选出正确的重组载体phy300p03,如附图3所示,分别为引物phy300
‑
f/r和引物yjhg
‑
f/yjhh
‑
r对载体phy300p03的pcr扩增验证,电泳结果与理论长度5175bp、2909bp相符,附图7为phy300p03的构建过程示意图。
[0099]
(11)将启动子p
43
连入载体php13。将载体进行hindiii和sali双酶切反应,用无缝克隆连接体系将p
43
启动子基因片段与线性化载体php13连接。
[0100]
(12)载体php13p01的筛选。将连接产物转化入大肠杆菌dh5α感受态中,涂布于含100μg/ml的氯霉素抗性的lb固体培养基(蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l,琼脂20g/l),37℃培养16
‑
18小时,挑取转化子进行菌落pcr验证,验证片段与理论片段相符合,即筛选出正确的重组载体php13p01,如附图4,为载体php13p01的hindiii
‑
ecori双酶切和php13
‑
f/r扩增验证,电泳结果与理论长度4713bp、344bp、889bp相一致。
[0101]
(13)将基因aceb、mdh基因连入载体php13p01。将载体进行xhoi和ecori双酶切反应,用无缝克隆连接体系将aceb和mdh连入线性化的质粒php13p01。
[0102]
(14)载体php13p02的筛选。将连接产物转化入大肠杆菌dh5α感受态中,涂布于含100μg/ml的氯霉素抗性的lb固体培养基(蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l,琼脂20g/l),37℃培养16
‑
18小时,挑取转化子进行菌落pcr验证,验证片段与理论片段相符合,即筛选出正确的重组载体php13p02,如附图5,为质粒载体的xhoi单酶切以及引物php13
‑
f/r,aceb
‑
f/mdh
‑
r的pcr扩增验证,电泳结果与理论序列长度7592bp、3424bp、2582bp均一致。
[0103]
(15)将基因alda基因连入载体php13p02。将载体进行sphi和xhoi双酶切反应,用无缝克隆连接体系将alda连入线性化的质粒php13p02。
[0104]
(16)载体php13p03的筛选。将连接产物转化入大肠杆菌dh5α感受态中,涂布于含100μg/ml的氯霉素抗性的lb固体培养基(蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l,琼脂20g/l),37℃培养16
‑
18小时,挑取转化子进行菌落pcr验证,验证片段与理论片段相符合,即筛选出正确的重组载体php13p03,如附图6所示,引物php13
‑
f/r对载体的pcr扩增验证,电泳结果与理论长度4640bp一致,附图8为php1303的构建过程示意图。
[0105]
实施例二:重组载体phy300p03和php13p03的电击转化bsu00
[0106]
(1)枯草芽孢杆菌的电转感受态制备
[0107]
将保存于
‑
80℃的枯草芽孢杆菌固体lb平板划线培养活化16h,挑取单菌落接种于50ml的液体lb培养基,220rpm,37℃条件下过夜培养;按照4%的接种量接种于新鲜的lbs生长培养基,37℃,220rpm条件下培养至od600为0.85到0.95之间;收集菌液于50ml干净灭菌的离心管中,在冰上预冷30min;6000rpm,4℃离心10min,倒掉上清液;用20ml洗涤培养基洗涤并重悬菌体,注意吹吸时要缓慢轻柔,6000rpm,4℃离心10min,倒掉上清液;重复上一步2
‑
3次;用1ml的电转重悬培养基重悬得到的菌体,并以每个100μl分装与干净无菌的1.5ml的离心管中,
‑
80℃保存备用;
[0108]
(2)载体phy300p03的电转化筛选
[0109]
将感受态置于冰上,在无菌环境下每100μl感受态添加4
‑
6μl的质粒,根据提取得到的质粒浓度灵活调整;电转杯在冰上预冷,将已经添加质粒的感受态转移到电转杯中;设置电转仪的参数,电压1.8kv至2.5kv,电击时间一般4.5.ms到5.ms之间;电击结束后立即加入37℃预热的复苏培养基900μl,220rpm,37℃培养3小时;5000rpm离心3min,留200μl培养基重悬菌体,加入到添加四环素抗生素的固体培养基,涂布棒涂板;37℃培养箱中培养,筛选转化子;
[0110]
(3)载体php13p03和phy300p03的转化子筛选
[0111]
将感受态置于冰上,在无菌环境下每100μl感受态添加4
‑
6μl的质粒,根据提取得到的质粒浓度灵活调整;电转杯在冰上预冷,将已经添加质粒的感受态转移到电转杯中;设置电转仪的参数,电压1.8kv至2.5kv,电击时间一般4.5.ms到5.ms之间;电击结束后立即加入37℃预热的复苏培养基900μl,220rpm,37℃培养3小时;5000rpm离心3min,留200μl培养基重悬菌体,加入到添加四环素和氯霉素抗生素的固体培养基,涂布棒涂板;37℃培养箱中培养,筛选转化子,如附图9,为两组模块下的代谢示意图;
[0112]
实施例三:重组菌株bsu02木糖为碳源产fengycin的发酵实验
[0113]
(1)培养基成分:
[0114]
固体培养基:蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l,琼脂20g/l;
[0115]
种子培养基:蛋白胨10g/l,酵母粉5g/l,氯化钠10g/l;
[0116]
发酵培养基:木糖15g/l,黄豆饼粉21.9g/l,nano
3 3g/l,mnso
4 0.2g/l;
[0117]
(2)发酵实验:将本发明构建的菌株bsu02在固体培养基中37℃培养活化20h,挑取单菌落于装有50ml种子培养基的250ml的摇瓶中,37℃,220rpm过夜培养,按5%的接种量取发酵种子液到发酵培养基中,30℃,200rpm的条件培养60h,发酵结束。
[0118]
(3)fengycin的提取和测定:取发酵液20ml于50ml的离心管中,在8000rpm,4℃离心30min,弃去菌体沉淀,在上清中加入6m的hcl调节ph至2.0,沉淀发酵液中游离的fengycin,4℃静置过夜,8000rpm,4℃离心30min,收集酸沉淀,然后用3ml纯甲醇溶剂萃取,萃取结束后用0.2m的naoh调节ph至7,离心收集上清液,即为含fengycin的粗提液。将发酵液处理所得的粗提液过0.22μm的滤膜,采用色谱柱4.6
×
150mm xdb c18进行hplc(agilent 1200,usa)分析,流动相为乙腈/水/三氟乙酸(50:50:0.5,v/v/v),柱温30℃,流速0.8ml/min,紫外检测波长为210nm,仪器的进样量为20μl。发酵产量的结果显示利用本发明提出的方法构建的利用dahms模块和调控模块乙醇醛回补tca模块组合表达的利用木糖合成fengycin的菌株bsu02的fengycin发酵产量达到46.82mg/l。
[0119]
本发明提出的工程菌株bsu02的构建方法为利用木糖提高fengycin的产量提供了一种新的策略。
[0120]
本发明公开和提出的技术方案,本领域技术人员可通过借鉴本文内容,适当改变条件路线等环节实现,尽管本发明的方法和制备技术已通过较佳实施例子进行了描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和技术路线进行改动或重新组合,来实现最终的制备技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,他们都被视为包括在本发明精神、范围和内容中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1