一种苯唑草酮的制备方法与流程
1.本发明涉及化学化工技术领域,特别涉及基于一氧化碳插羰法的一种苯唑草酮的制备方法。
背景技术:
2.苯唑草酮(topramezon)是德国巴斯夫公司开发的一种苯甲酯吡唑酮类除草剂,该化合物属于三酮类苗后茎叶处理剂,通过抑制质体醌生物合成中的4
‑
羟基苯基丙酮酸酯双氧化酶(4
‑
hppd),间接影响类胡萝卜素的合成,从而干扰叶绿素的合成和功能,最终导致严重的白化。此类化合物对于杂草有很好的防除效果,能有效防除世界范围内玉米作物上的主要禾本科杂草和阔叶杂草。苯唑草酮具有杀草谱广、活性高、可混性强,用量少以及对玉米和后茎作物安全等优点,是一类比较安全的除草剂。目前,如何高效合成该类化合物是国内相继研发的重点领域。由于巴斯夫研发的专利路线中使用了昂贵的钯类催化剂造成了其成本极为昂贵,并且钯催化剂回收困难,造成在大规模生产过程中整个生产成本消耗过高。因此,探索使用新的廉价金属催化剂催化该反应,从根本上降低生产成本,解决苯唑草酮价格昂贵的问题,为国内提供廉价高效的苯唑草酮产品是十分紧迫的任务。
技术实现要素:
3.为了克服上述现有技术的缺陷,本发明的目的在于提供一种苯唑草酮的制备方法,具有高效、价格低廉、绿色环保的特点。
4.为达到上述目的,本发明采用如下技术方案:
5.一种苯唑草酮的制备方法,其特征在于,包括以下步骤:
6.将化合物ⅰ(3
‑
[3
‑
溴
‑2‑
甲基
‑6‑
(甲硫基)
‑
苯基]
‑
4,5
‑
二氢异噁唑)和化合物ⅱ(1
‑
甲基
‑5‑
羟基吡唑)在铜类催化剂、配体、有机溶剂、碱和co气体氛围中进行反应制备苯唑草酮,控制反应温度为25~120℃;反应时长为18~24小时。
[0007]
所述化合物ⅰ的结构式为:所述化合物ⅱ的结构式为:所述苯唑草酮的结构式为:
[0008]
所述的铜类催化剂为氯化亚铜、溴化亚铜、碘化亚铜、醋酸铜、硫酸铜、乙酰丙酮铜,选用其中任意一种或多种任意比例混合。
[0009]
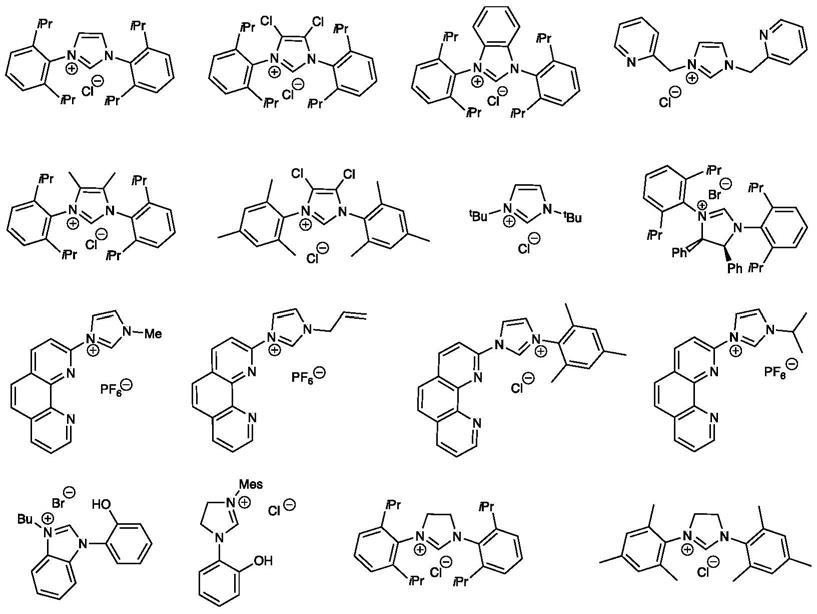
所述的配体为氮杂环卡宾类配体,具体结构如下图所示,
[0010][0011]
所述的有机溶剂为选自四氢呋喃,dmso,乙睛,1,4
‑
二氧六环,甲苯,n,n
‑
二甲基甲酰胺、n
‑
甲基吡咯烷酮中的一种或多种任意比例。
[0012]
所述的碱为选自碳酸钠、碳酸钾、碳酸铯、碳酸氢钾、碳酸氢钠、叔丁醇锂、叔丁醇钠、叔丁醇钾、甲醇钠、甲醇钾、乙醇钠、乙醇钾、三乙胺、三乙烯二胺、4
‑
二甲氨基吡啶、二乙丙基胺基锂中的一种或多种任意比例。
[0013]
所述的co气体,通入时控制反应釜内的气体压力为1~30bar。
[0014]
所述的化合物ⅰ和所述的化合物ⅱ的投料摩尔比为1:(1~2.5)。所述的铜类催化剂与配体投料比为1:1~2。
[0015]
所述化合物ⅰ和铜类催化剂的摩尔比为1:0.01~0.1;所述化合物ⅰ与碱的摩尔比为1:1~4。
[0016]
所述的化合物ⅰ和化合物ⅱ需要在40~100℃烘箱中烘干除去化合物中的水分。
[0017]
进一步优化:
[0018]
所述化合物ⅰ和化合物ⅱ的投料摩尔比为1:(1.1~1.4)。
[0019]
所述铜类催化剂与配体投料比为1:(1~1.1);或者铜类催化剂和配体预先制成卡宾铜催化剂,用于催化反应,其中预制阶段催化剂与配体投料比也为1:(1~1.1)。
[0020]
所述化合物ⅰ与铜类催化剂的摩尔比为1:(0.05~0.1)。
[0021]
所述化合物ⅰ与碱的摩尔比为1:2~4。
[0022]
所述的苯唑草酮的制备方法,具体为:
[0023]
将所述的化合物ⅰ和所述的化合物ⅱ溶于所述的有机溶剂中,加入所述的铜类催化剂与配体或者已经预制好的卡宾铜催化剂,再加入所述的碱,通入co气体置换釜内空气3次,每次置换时间不少于10分钟,于100~120℃下反应18~24小时,得到的反应混合物自然
冷却至室温,使用真空旋转蒸发仪出除去低沸点物质,然后加入质量分数1.2的%naoh溶液调节ph=12,抽滤过滤出溶液中的不溶物,滤液用二氯甲烷萃取一遍,分出水层,有机层再用质量分数1.2%的naoh溶液萃取一遍,合并水层,用质量分数36%盐酸调节水相的ph=2~3,大量黄色固体析出,抽滤得到黄色固体,随后将固体置于40℃烘箱中烘干,所得即为苯唑草酮。
[0024]
本发明的反应方程式如下:
[0025][0026]
由于上述方案运用,本发明与现有技术相比具有下列优势:
[0027]
本发明使用的工艺相对简单,避免应用贵金属钯类催化剂,反应过程所用试剂和药品以及反应条件的成本相对较低,而且反应时间短,收率高,适宜工业化生产。
附图说明
[0028]
图1是苯唑草酮核磁数据氢谱图。
[0029]
图2是苯唑草酮核磁数据碳谱图。
具体实施方式
[0030]
下面结合实例对本发明做进一步描述。但本发明并不限于以下实施例。实施例中采用的实施条件可以根据具体使用的不同要求做进一步调整,未注明的实验条件为本行业中的常规条件。本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互结合。
[0031]
实施例一
[0032]
一种苯唑草酮的制备方法,包括以下步骤:
[0033]
将20g(62.8mmol)化合物ⅰ(3
‑
[3
‑
溴
‑2‑
甲基
‑6‑
(甲硫基)
‑
苯基]
‑
4,5
‑
二氢异噁唑)和化合物ⅱ(1
‑
甲基
‑5‑
羟基吡唑)6.78g(69.0mmol)完全溶于1,4
‑
二氧六环中,加入催化剂氯化亚铜0.314g(3.14mmol),配体1,3
‑
双(2,6
‑
二异丙基苯基)咪唑
‑2‑
亚基1.218g(3.14mmol),三乙胺15.9g(0.157mol),25barco置换反应釜内的空气三次,每次置换时间10min,3次置换完成后在100℃下反应18小时;反应结束后自然冷却至室温,得到的反应混合物使用真空旋转蒸发仪出除去三乙胺和1,4
‑
二氧六环,然后加入质量分数1.2%的naoh的溶液调节ph=12,抽滤过滤出溶液中的不溶物,滤液用二氯甲烷萃取一遍,分出水层,有机层再用质量分数1.2%的naoh的溶液萃取一遍,合并水层,用质量分数36%盐酸调节水相的ph=2~3,有大量黄色固体析出,抽滤得到黄色固体,随后将固体置于40℃烘箱中烘干,得到产品20.5g,纯度为95%,收率为89.8%。产物结构经过核磁数据表征,从产物氢谱图1和碳谱图2中可以确定得到目标产物为苯唑草酮,即结构和化学式分别为:
[0034]
(3
‑
(4,5
‑
dihydroisoxazol
‑3‑
yl)
‑2‑
methyl
‑4‑
(methylsulfonyl)phenyl)(5
‑
hydroxy
‑1‑
methyl
‑
1h
‑
pyrazol
‑4‑
yl)methanone;
[0035]
氢谱为:
[0036]1h nmr(400mhz,dmso
‑
d6)δ7.93(d,j=8.4hz,1h),7.63(d,j=8.4hz,1h),7.34(s,1h),4.43(t,j=10.0hz,2h),3.51(s,3h),3.31(t,j=10.0hz,2h),3.22(s,3h),2.17(s,3h);
[0037]
碳谱为:
[0038]
13
c nmr(100mhz,dmso
‑
d6)δ188.44,157.52,155.54,146.08,140.89,140.52,136.38,130.57,128.67,127.57,105.53,69.49,45.54,40.47,33.56,16.66。
[0039]
其余实施例与实施例一步骤一致,唯一改变的是某些反应参数,
[0040]
具体参数调整对反应收率的影响见下表:
[0041]
[0042]
[0043][0044]
以上对本发明做了详尽的描述,其目的在于让熟悉此领域技术的人士能够了解本发明的内容并加以实施,并不能因此限制本发明的保护范围,凡根据本发明的精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1