一种美罗培南侧链光学异构体及其制备方法和应用、美罗培南侧链杂质的检测方法与流程
1.本发明涉及药物中间体合成技术领域,尤其涉及一种美罗培南侧链光学异构体及其制备方法和应用、美罗培南侧链杂质的检测方法。
背景技术:
2.美罗培南(meropenem)属于碳青霉烯类抗生素,对厌氧和需氧的革兰氏阳性菌、阴性菌等有良好的抗菌作用,尤其对多重耐药的需氧革兰氏阴性杆菌仍具有抗菌活性。由于化学结构在碳青霉烯母环的4位上引入了甲基,其对人的肾去氢肽酶-1(dhp-1)的稳定性明显增高,因此不需要同时应用 dhp-1抑制剂,如西司他丁,可以单独进行临床使用。美罗培南侧链 (meropenem side chain,简称2s4s-m),即(2s,4s)-2-二甲基氨基甲酰-4-巯基-1-(对硝基苄氧羰基)-1-吡咯烷(如下式1所示),是生产碳青霉烯类抗生素美罗培南的关键中间体,该化合物含有两个手性中心。
[0003][0004]
随着世界各国对药品杂质危害性的认识越来越深刻,对药物中间体的质量要求也越来越高,美罗培南侧链作为关键医药中间体,其质量情况及纯度直接影响药品的制备及其安全性。由于手性药物及其异构体在人体内可能会产生不同甚至相反的药理作用,因此对合成美罗培南的关键中间体-美罗培南侧链进行光学异构体的合成具有重要的意义。
技术实现要素:
[0005]
本发明的目的在于提供一种美罗培南侧链光学异构体及其制备方法和应用、美罗培南侧链杂质的检测方法,所述美罗培南侧链光学异构体作为美罗培南侧链的杂质,能够作为标准品实现对美罗培南侧链中杂质的检测。
[0006]
为了实现上述发明目的,本发明提供以下技术方案:
[0007]
本发明提供了一种具有式i所示结构通式的美罗培南侧链光学异构体,
[0008][0009]
所述美罗培南侧链光学异构体具有式ii、式iii或式iv所示结构构型:
[0010][0011]
本发明提供了上述技术方案所述美罗培南侧链光学异构体的制备方法,包括以下步骤:
[0012]
当所述美罗培南侧链光学异构体具有式ii所示结构构型时,所述美罗培南侧链光学异构体的制备方法包括以下步骤:
[0013]
将顺式-d-羟脯氨酸、氯甲酸对硝基苄酯、第一溶剂、氢氧化钠和水混合,进行酰胺化反应,得到中间体1;
[0014]
将所述中间体1、三乙胺、第二溶剂、氯甲酸异丙酯和二甲胺混合,进行酰胺化反应,得到中间体2;
[0015]
将所述中间体2、三乙胺、第三溶剂与甲磺酰氯混合,进行磺酰化反应,得到中间体3;
[0016]
将所述中间体3、硫代醋酸钾和第四溶剂混合,进行硫代反应,得到中间体4;
[0017]
将所述中间体4与碱液混合,进行水解,得到具有式ii所示结构构型的美罗培南侧链光学异构体;
[0018]
所述中间体1具有式a所示结构:
[0019]
所述中间体2具有式b所示结构:
[0020]
所述中间体3具有式c所示结构:
[0021]
所述中间体4具有式d所示结构:
[0022]
当所述美罗培南侧链光学异构体具有式iii所示结构构型时,所述美罗培南侧链光学异构体的制备方法包括以下步骤:
[0023]
将所述中间体3、溴化钠、碘化钾和第五溶剂混合,进行溴代反应,得到中间体5;
[0024]
将所述中间体5与硫代醋酸钾混合,进行取代反应,得到中间体6;
[0025]
将所述中间体6与碱液混合,进行水解,得到具有式iii所示结构构型的美罗培南侧链光学异构体;
[0026]
所述中间体5具有式e所示结构:
[0027]
所述中间体6具有式f所示结构:
[0028]
当所述美罗培南侧链光学异构体具有式iv所示结构构型时,所述美罗培南侧链光学异构体的制备方法包括以下步骤:
[0029]
将反式-l-羟脯氨酸、氯甲酸对硝基苄酯、第六溶剂、氢氧化钠和水混合,进行酰胺化反应,得到中间体7;
[0030]
将所述中间体7、三乙胺、第七溶剂和氯甲酸异丙酯混合,进行酯化反应,将所得产物与甲磺酰氯混合,进行磺酰化反应,得到中间体8;
[0031]
将所述中间体8与二甲胺混合,进行酰胺化反应,得到中间体9;
[0032]
将所述中间体9、溴化钠、碘化钾和第八溶剂混合,进行溴代反应,得到中间体10;
[0033]
将所述中间体10与硫代醋酸钾混合,进行取代反应,得到中间体11;
[0034]
将所述中间体11与碱液混合,进行水解,得到具有式iv所示结构构型的美罗培南侧链光学异构体;
[0035]
所述中间体7具有式j所示结构:
[0036]
所述中间体8具有式h所示结构:
[0037]
所述中间体9具有式i所示结构:
[0038]
所述中间体10具有式g所示结构:
[0039]
所述中间体11具有式k所示结构:
[0040]
优选的,制备具有式ii所示结构构型的美罗培南侧链光学异构体时,所所述顺式-d-羟脯氨酸和氯甲酸对硝基苄酯的摩尔比为(0.76:0.86;制备所述中间体1时,所述(0.6~1.0):(0.8~1.2);所述酯化反应的温度为-5~10℃,时间为1~6h。
[0041]
优选的,制备中间体2时,所述中间体1、三乙胺、氯甲酸异丙酯和二甲胺的摩尔比为(0.10~0.15):(0.15~0.2):(0.12~0.18):(0.12~0.2);所述酰胺化反应的温度为-5~10℃,时间为1~6h;
[0042]
制备中间体3时,所述中间体2、三乙胺与甲磺酰氯的摩尔比为 (0.10~0.15):(0.13~0.25):(0.11~0.2);所述磺酰化反应的温度为-5~10℃,时间为1~6h。
[0043]
优选的,制备中间体4时,所述中间体3和硫代醋酸钾的摩尔比为 (0.010~0.015):(0.022~0.028),所述硫代反应的温度为40~60℃,时间为 2~8h。
[0044]
优选的,制备中间体5时,所述中间体3、溴化钠和碘化钾的摩尔比为 (0.04~0.05):(0.20~0.03):(0.001~0.01);所述溴代反应的温度为70~120℃,时间为1~5h。
[0045]
优选的,制备中间体7时,所述反式-l-羟脯氨酸和氯甲酸对硝基苄酯的摩尔比为(0.6~0.8):(0.8~1.0);所述酰胺化反应的温度为-5~10℃,时间为1~6h;
[0046]
制备中间体8时,所述中间体7、三乙胺、氯甲酸异丙酯和甲磺酰氯的摩尔比为(0.10~0.15):(0.2~0.4):(0.1~0.16):(0.11~0.2);所述酯化反应的温度为-5~10℃,时间为1~6h;
[0047]
制备中间体9时,所述中间体8与二甲胺的摩尔比为(0.1~0.2):(0.1~0.3)。
[0048]
本发明提供了上述技术方案所述美罗培南侧链光学异构体或上述技术方案所述制备方法制备得到的美罗培南侧链光学异构体作为标准品在检测美罗培南侧链产品杂质中的应用。
[0049]
本发明提供了一种美罗培南侧链杂质的检测方法,包括以下步骤:
[0050]
采用高效液相色谱法,以美罗培南侧链光学异构体作为杂质标准品,进行液相色谱检测,得到标准液相色谱谱图;所述美罗培南侧链光学异构体为上述技术方案所述美罗培南侧链光学异构体或上述技术方案所述制备方法制备得到的美罗培南侧链光学异构体;
[0051]
以美罗培南侧链样品作为待测样品,进行液相色谱检测,得到待测样品液相色谱谱图;
[0052]
将所述待测样品液相色谱谱图与标准液相色谱谱图进行对比,按照面积归一化法计算待测样品中杂质含量。
[0053]
优选的,所述液相色谱检测的色谱条件为:色谱柱:chiralpak ic,250 mm
×
4.6mm,5μm;流动相:正己烷、乙醇、甲醇和三氟乙酸的混合物,所述正己烷、乙醇、甲醇和三氟乙酸的体积比为(500~700):(200~400):(50~200): (1~10);流速:1.0~1.5ml/min;检测波长:270nm;进样量:20~25μl;柱温:30~60℃;采集时间:20~50min。
[0054]
本发明提供了一种美罗培南侧链光学异构体,本发明提供的美罗培南侧链光学异构体为美罗培南侧链生产过程中影响美罗培南侧链产品纯度的物质,即所产生的杂质,所述美罗培南侧链光学异构体能够作为标准品实现对美罗培南侧链中杂质的有效检测,为提高美罗培南侧链产品的质量奠定了基础。
附图说明
[0055]
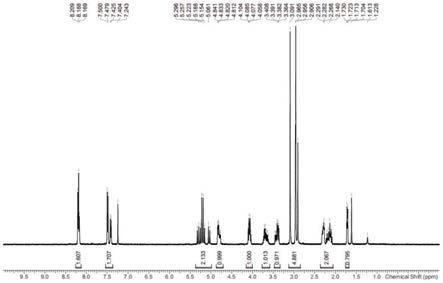
图1为实施例1制备的产物的核磁谱图;
[0056]
图2为实施例1制备的产物的质谱图;
[0057]
图3为实施例2制备的产物的核磁谱图;
[0058]
图4为实施例2制备的产物的质谱图;
[0059]
图5为实施例3制备的产物的核磁谱图;
[0060]
图6为实施例3制备的产物的质谱图;
[0061]
图7为美罗培南侧链的手性液相谱图;
[0062]
图8为美罗培南侧链光学异构体2s4r-m手性纯度谱图;
[0063]
图9为美罗培南侧链光学异构体2r4s-m手性纯度谱图;
[0064]
图10为美罗培南侧链光学异构体2r4r-m手性纯度谱图;
[0065]
图11为美罗培南侧链与三个异构体混合样品谱图。
具体实施方式
[0066]
本发明提供了一种具有式i所示结构通式的美罗培南侧链光学异构体,
[0067][0068]
所述美罗培南侧链光学异构体具有式ii、式iii或式iv所示结构构型:
[0069][0070]
本发明提供了上述技术方案所述美罗培南侧链光学异构体的制备方法,包括以下步骤:
[0071]
当所述美罗培南侧链光学异构体具有式ii所示结构构型时,所述美罗培南侧链光学异构体的制备方法包括以下步骤:
[0072]
将顺式-d-羟脯氨酸、氯甲酸对硝基苄酯、第一溶剂、氢氧化钠和水混合,进行酰胺化反应,得到中间体1;
[0073]
将所述中间体1、三乙胺、第二溶剂、氯甲酸异丙酯和二甲胺混合,进行酰胺化反应,得到中间体2;
[0074]
将所述中间体2、三乙胺、第三溶剂与甲磺酰氯混合,进行磺酰化反应,得到中间体3;
[0075]
将所述中间体3、硫代醋酸钾和第四溶剂混合,进行硫代反应,得到中间体4;
[0076]
将所述中间体4与碱液混合,进行水解,得到具有式ii所示结构构型的美罗培南侧链光学异构体;
[0077]
所述中间体1具有式a所示结构:
[0078]
所述中间体2具有式b所示结构:
[0079]
所述中间体3具有式c所示结构:
[0080]
所述中间体4具有式d所示结构:
[0081]
当所述美罗培南侧链光学异构体具有式iii所示结构构型时,所述美罗培南侧链光学异构体的制备方法包括以下步骤:
[0082]
将所述中间体3、溴化钠、碘化钾和第五溶剂混合,进行溴代反应,得到中间体5;
[0083]
将所述中间体5与硫代醋酸钾混合,进行取代反应,得到中间体6;
[0084]
将所述中间体6与碱液混合,进行水解,得到具有式iii所示结构构型的美罗培南侧链光学异构体;
[0085]
所述中间体5具有式e所示结构:
[0086]
所述中间体6具有式f所示结构:
[0087]
当所述美罗培南侧链光学异构体具有式iv所示结构构型时,所述美罗培南侧链光学异构体的制备方法包括以下步骤:
[0088]
将反式-l-羟脯氨酸、氯甲酸对硝基苄酯、第六溶剂、氢氧化钠和水混合,进行酰胺化反应,得到中间体7;
[0089]
将所述中间体7、三乙胺、第七溶剂和氯甲酸异丙酯混合,进行酯化反应,将所得产物与甲磺酰氯混合,进行磺酰化反应,得到中间体8;
[0090]
将所述中间体8与二甲胺混合,进行酰胺化反应,得到中间体9;
[0091]
将所述中间体9、溴化钠、碘化钾和第八溶剂混合,进行溴代反应,得到中间体10;
[0092]
将所述中间体10与硫代醋酸钾混合,进行取代反应,得到中间体11;
[0093]
将所述中间体11与碱液混合,进行水解,得到具有式iv所示结构构型的美罗培南侧链光学异构体;
[0094]
所述中间体7具有式j所示结构:
[0095]
所述中间体8具有式h所示结构:
[0096]
所述中间体9具有式i所示结构:
[0097]
所述中间体10具有式g所示结构:
[0098]
所述中间体11具有式k所示结构:
[0099]
在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
[0100]
在本发明中,当所述美罗培南侧链光学异构体具有式ii所示结构构型时:
[0101]
本发明将顺式-d-羟脯氨酸、氯甲酸对硝基苄酯、第一溶剂、氢氧化钠和水混合,进行酰胺化反应,得到中间体1。在本发明中,所述顺式-d-羟脯氨酸和氯甲酸对硝基苄酯的摩尔比优选为(0.6~0.8):(0.8~1.0),更优选为 0.76:0.86。在本发明中,所述第一溶剂优选为二氯甲烷,所述二氯甲烷用于溶解氯甲酸对硝基苄酯,所述氯甲酸对硝基苄酯的二氯甲烷溶液的质量浓度优选为50%;所述氢氧化钠与水的质量比优选为66.9:600;所述氢氧化钠与顺式-d-羟脯氨酸的摩尔比优选为1.68:0.76。
[0102]
在本发明中,所述顺式-d-羟脯氨酸、氯甲酸对硝基苄酯、第一溶剂、氢氧化钠和水混合的过程优选为先将氯甲酸对硝基苄酯溶解于第一溶剂,得到氯甲酸对硝基苄酯溶液,将氢氧化钠和水搅拌溶解后,再加入顺式-d-羟脯氨酸,搅拌全溶后降温到0℃,滴加氯甲酸
对硝基苄酯的二氯甲烷溶液(控制温度不超过5℃)。本发明对所述搅拌、滴加和降温的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0103]
在本发明中,所述酯化反应的温度优选为-5~10℃,更优选为0~5℃,时间优选为1~6h,更优选为3h。
[0104]
完成所述酰胺化反应后,本发明优选将所得物料进行后处理,所述后处理的过程优选包括将所得物料进行静置分层,将所得水相用二氯甲烷提杂,分出水相,用质量分数为10%的盐酸调ph值至2~3,然后采用乙酸乙酯萃取三次,合并有机相,依次使用饱和盐水洗、无水硫酸钠干燥、过滤、浓缩至干和乙醇重结晶,得到中间体1。本发明对上述后处理过程的具体操作方法没有特殊的限定,按照本领域熟知的过程进行即可。
[0105]
在本发明中,所述中间体1具有式a所示结构:
[0106]
得到中间体1后,本发明将所述中间体1、三乙胺、第二溶剂、氯甲酸异丙酯和二甲胺混合,进行酰胺化反应,得到中间体2。在本发明中,所述中间体1、三乙胺、氯甲酸异丙酯和二甲胺的摩尔比优选为 (0.10~0.15):(0.15~0.2):(0.12~0.18):(0.12~0.2),更优选为0.13:0.19:0.15:0.18;所述第二溶剂优选为二氯甲烷;本发明对所述第二溶剂的用量没有特殊的限定,能够保证反应顺利进行即可。在本发明中,所述二甲胺优选以二甲胺的水溶液的形式使用,所述二甲胺的水溶液的质量浓度优选为40%。
[0107]
在本发明中,所述中间体1、三乙胺、第二溶剂、氯甲酸异丙酯和二甲胺混合的过程优选为将中间体1、三乙胺和二氯甲烷搅拌至全溶,降温至
ꢀ‑
10℃,滴加氯甲酸异丙酯,搅拌2h,滴加二甲胺水溶液(控制温度不超过 0℃)。本发明对所述滴加的速率没有特殊的限定,按照本领域熟知的速率滴加即可。
[0108]
在本发明中,所述酰胺化反应优选在氮气保护条件下进行,所述酰胺化反应的温度优选为-5~10℃,更优选为0℃,时间优选为1~6h,更优选为3h。
[0109]
完成所述酰胺化反应后,本发明优选向所得物料中加入水,搅拌5分钟后静置分层,将所得有机相依次用质量分数2%的稀盐酸洗、质量分数5%的碳酸钾水溶液洗、水洗和饱和食盐水洗,将所得洗涤物料分出有机层,将所得有机层物料使用无水硫酸钠干燥,过滤,浓缩至干,采用甲醇重结晶,得到中间体2。本发明对所加入水的量没有特殊的限定,根据实际需求进行调整即可。
[0110]
在本发明中,所述中间体2具有式b所示结构:
[0111]
得到中间体2后,本发明将所述中间体2、三乙胺、第三溶剂与甲磺酰氯混合,进行磺酰化反应,得到中间体3。在本发明中,所述中间体2、三乙胺与甲磺酰氯的摩尔比优选为(0.10~0.15):(0.13~0.25):(0.11~0.2),更优选为0.14:0.19:0.15;所述第三溶剂优选
为二氯甲烷;本发明对所述第三溶剂的用量没有特殊的限定,能够保证反应顺利进行即可。
[0112]
在本发明中,所述中间体2、三乙胺、第三溶剂与甲磺酰氯混合的过程优选为将中间体2、三乙胺和二氯甲烷搅拌至全溶,降温至-10℃,滴加甲磺酰氯(控制温度不超过0℃)。
[0113]
在本发明中,所述磺酰化反应优选在氮气保护条件下进行,所述磺酰化反应的温度优选为-5~10℃,更优选为0℃,时间优选为1~6h,更优选为2h。
[0114]
完成所述磺酰化反应后,本发明优选向所得物料中加入水,搅拌10分钟后静置分层,将所得有机相依次用质量分数为2%的稀盐酸洗,质量分数 5%的碳酸钾洗、水洗和饱和食盐水洗,分出有机层,使用无水硫酸钠干燥,过滤,浓缩至干,用甲醇重结晶,得到中间体3。本发明对所加入水的量没有特殊的限定,根据实际需求进行调整即可。
[0115]
在本发明中,所述中间体3具有式c所示结构:
[0116]
得到中间体3后,本发明将所述中间体3、硫代醋酸钾和第四溶剂混合,进行硫代反应,得到中间体4。在本发明中,所述中间体3和硫代醋酸钾的摩尔比优选为(0.010~0.015):(0.022~0.028),更优选为0.013:0.026;所述第四溶剂优选为n,n-二甲基甲酰胺;本发明对所述第四溶剂的用量没有特殊的限定,能够保证反应顺利进行即可。
[0117]
本发明对所述中间体3、硫代醋酸钾和第四溶剂混合的过程没有特殊的限定,按照本领域熟知的过程能够将物料充分混合即可。
[0118]
在本发明中,所述硫代反应优选在氮气保护条件下进行,所述硫代反应的温度优选为40~60℃,更优选为55℃,时间优选为2~8h,更优选为3h。
[0119]
完成所述硫代反应后,本发明优选将所得物料降温至室温,加入水,搅拌10分钟,然后用乙酸乙酯提取三次,合并有机层,依次采用水洗3次和饱和食盐水洗3次,分出有机层,将所得有机层物料依次进行无水硫酸钠干燥、过滤和浓缩至干,得到中间体4。本发明对所述加入水的用量没有特殊的限定,根据实际需求进行调整即可。
[0120]
在本发明中,所述中间体4具有式d所示结构:所述中间体4具有式d所示结构:
[0121]
得到中间体4后,本发明将所述中间体4与碱液混合,进行水解,得到具有式ii所示结构构型的美罗培南侧链光学异构体,记为2r4s-m。
[0122]
在本发明中,所述中间体4与碱液中碱的摩尔比优选为0.013:0.03;所述碱液优选为氢氧化钾的水溶液,本发明对所述碱液的浓度没有特殊的限定,能够保证反应顺利进行即可。
[0123]
在所述中间体4与碱液混合的过程中,还需要加入二氯甲烷,本发明对所述二氯甲烷的用量没有特殊的限定,能够保证反应顺利进行即可。
[0124]
在本发明中,所述中间体4与碱液混合的过程优选为在氮气保护条件下,将中间体
4溶解于二氯甲烷,降温至0~5℃,滴加碱液25ml(温度不超过10℃)。
[0125]
在本发明中,所述水解优选在氮气保护条件下进行;所述水解的温度优选为0~10℃且不为0℃,时间优选为1.5h。
[0126]
完成所述水解后,本发明优选向所得物料加入水,降温至0℃,用质量分数10%的盐酸调ph至7(温度不超过5℃),将所得物料静置分液,将所得有机层水洗两次后再用饱和食盐水洗,分出有机层,无水硫酸钠干燥,过滤,浓缩,柱层析纯化(洗脱剂优选为体积比为2:1的乙酸乙酯和石油醚),得到2r4s-m。本发明对所述加入水的用量没有特殊的限定,根据实际需求进行调整即可。
[0127]
在本发明中,所述2r4s-m的合成过程如下式所示:
[0128][0129]
在本发明中,当所述美罗培南侧链光学异构体具有式iii所示结构构型时:
[0130]
本发明将所述中间体3、溴化钠、碘化钾和第五溶剂混合,进行溴代反应,得到中间体5。在本发明中,所述中间体3、溴化钠和碘化钾的摩尔比优选为(0.04~0.05):(0.20~0.03):(0.001~0.01),更优选为0.048:0.241:0.001;所述第五溶剂优选为n,n-二甲基甲酰胺;本发明对所述第五溶剂的用量没有特殊的限定,能够保证反应顺利进行即可。
[0131]
本发明对所述中间体3、溴化钠、碘化钾和第五溶剂混合的过程没有特殊的限定,按照本领域熟知的过程能够将物料混合均匀即可。
[0132]
在本发明中,所述溴代反应优选在氮气保护条件下进行,所述溴代反应的温度优选为70~120℃,更优选为100℃,时间优选为1~5h,更优选为2h。
[0133]
完成所述溴代反应后,本发明优选将所得物料降温至室温,加入水,搅拌10分钟,然后用乙酸乙酯提取三次,合并有机层,依次采用水洗3次和饱和食盐水洗3次,分出有机层,无水硫酸钠干燥,过滤,浓缩至干,柱层析纯化,得到中间体5。在本发明中,所述柱层析所用洗脱剂优选为乙酸乙酯和石油醚的混合物,所述乙酸乙酯和石油醚的体积比优选为1:1。本发明对所述加入水的用量没有特殊的限定,根据实际需求进行调整即可。本发明对所述过滤和浓缩的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0134]
在本发明中,所述中间体5具有式e所示结构:
[0135]
得到中间体5后,本发明将所述中间体5与硫代醋酸钾混合,进行取代反应,得到中间体6。在本发明中,所述中间体5与硫代醋酸钾的摩尔比优选为(0.010~0.015):(0.022~0.028),更优选为0.013:0.026。
[0136]
在所述中间体5与硫代醋酸钾混合过程中,还包括加入溶剂;所述溶剂优选为n,n-二甲基甲酰胺;本发明对所述中间体5、硫代醋酸钾和溶剂混合的过程没有特殊的限定,按照本领域熟知的过程能够将物料充分混合即可;本发明对所述溶剂的用量没有特殊的限定,保证反应顺利进行即可。
[0137]
在本发明中,所述取代反应优选在氮气保护条件下进行,所述取代反应的温度优选为40~60℃,更优选为55℃;时间优选为2~8h,更优选为3h;
[0138]
完成所述取代反应后,本发明优选将所得物料进行后处理,具体过程优选为将所得粗品产物液降至室温,加入水,搅拌10分钟,然后用乙酸乙酯提取三次,合并有机层,依次进行水洗和饱和食盐水洗,分出有机层,将所得有机层物料采用无水硫酸钠干燥,过滤后,浓缩至干,得到中间体6。所述水洗的次数优选为3次,所述饱和食盐水洗的次数优选为3次本发明对所述加入水的用量没有特殊的限定,根据实际需求进行调整即可。本发明对所述干燥、过滤和浓缩的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0139]
在本发明中,所述中间体6具有式f所示结构:所述中间体6具有式f所示结构:
[0140]
得到中间体6后,本发明将所述中间体6与碱液混合,进行水解,得到具有式iv所示结构构型的美罗培南侧链光学异构体,记为2r4r-m。在本发明中,所述中间体6与碱液混合进行水解的过程优选与中间体4进行水解的过程完全相同,在此不在赘述。
[0141]
在本发明中,所述2r4r-m的合成过程如下式所示:
[0142][0143]
在本发明中,当所述美罗培南侧链光学异构体具有式iv所示结构构型时:
[0144]
本发明将反式-l-羟脯氨酸、氯甲酸对硝基苄酯、第六溶剂、氢氧化钠和水混合,进行酰胺化反应,得到中间体7。在本发明中,所述反式-l-羟脯氨酸和氯甲酸对硝基苄酯的摩尔比优选为(0.6~0.8):(0.8~1.0),更优选为 0.76:0.86;所述第六溶剂优选为二氯甲烷,所述二氯甲烷用于溶解氯甲酸对硝基苄酯,所述氯甲酸对硝基苄酯的二氯甲烷溶液的质量浓度优选为50%;所述氢氧化钠与水的质量比优选为66.9:600;所述氢氧化钠与反式-d-羟脯氨酸的摩尔比优选为1.68:0.76。
[0145]
在本发明中,所述反式-d-羟脯氨酸、氯甲酸对硝基苄酯、第六溶剂、氢氧化钠和水混合的过程优选为先将氯甲酸对硝基苄酯溶解于第六溶剂,得到氯甲酸对硝基苄酯溶液,将氢氧化钠和水搅拌溶解后,再加入顺式-d-羟脯氨酸,搅拌全溶后降温到0℃,滴加氯甲酸对硝基苄酯的二氯甲烷溶液(控制温度不超过5℃)。本发明对所述搅拌、滴加和降温的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0146]
在本发明中,所述酰胺化反应的温度优选为-5~10℃,更优选为0~5℃,时间优选为1~6h,更优选为3h。
[0147]
完成所述酰胺化反应后,本发明优选将所得物料进行后处理,所述后处理的过程优选包括将所得物料进行静置分层,将所得水相用二氯甲烷提杂,分出水相,用质量分数为10%的盐酸调ph值至2~3,然后采用乙酸乙酯萃取三次,合并有机相,依次使用饱和盐水洗、无水硫酸钠干燥、过滤、浓缩至干和乙醇重结晶,得到中间体7。本发明对上述后处理过程的具体操作方法没有特殊的限定,按照本领域熟知的过程进行即可。
[0148]
在本发明中,所述中间体7具有式j所示结构:
[0149]
得到中间体7后,本发明将所述中间体7、三乙胺、第七溶剂和氯甲酸异丙酯混合,进行酰胺化反应,将所得产物与甲磺酰氯混合,进行磺酰化反应,得到中间体8。在本发明中,所述中间体7、三乙胺、氯甲酸异丙酯和甲磺酰氯的摩尔比优选为(0.10~0.15):(0.2~0.4):(0.1~0.16):(0.11~0.2),更优选为0.1:0.28:0.11:0.13;所述第七溶剂优选为二氯甲烷,本发明对所述第七溶剂的用量没有特殊的限定,能够保证反应顺利进行即可。
[0150]
在本发明中,所述中间体7、三乙胺、第七溶剂和氯甲酸异丙酯混合的过程优选为将中间体7、三乙胺和二氯甲烷混合,降温至-10℃,滴加氯甲酸异丙酯(温度不超过0℃)。在本发明中,所述酯化反应优选在氮气保护条件下进行,反应的温度优选为-5~10℃,更优选为0℃,时间优选为1~6h,更优选为2h。
[0151]
在本发明中,将所得产物与甲磺酰氯混合的过程优选为将反应所得物料降温至-10℃,滴加甲磺酰氯(温度不超过0℃);所述磺酰化反应优选在氮气保护条件下进行,所述磺酰化反应的温度优选为0℃,时间优选为2h。
[0152]
在本发明中,所述中间体8具有式h所示结构:
[0153]
得到中间体8后,本发明优选不进行任何后处理,直接向所得物料中滴加二甲胺,进行酰胺化反应,得到中间体9。在本发明中,所述二甲胺优选以二甲胺的水溶液的形式使用,所述二甲胺的水溶液的质量浓度优选为.40%;所述中间体8与二甲胺的摩尔比优选为(0.1~0.2):(0.1~0.3),更优选为0.1:0.13。本发明对所述滴加的速率没有特殊的限定,按照本领域熟知的速率滴加即可;所述滴加过程中,温度不超过10℃。在本发明中,所述酰胺化反应优选在氮气保护条件下进行,反应的温度优选为0~10℃且不为0,时间优选为3h。
[0154]
完成所述酰胺化反应后,优选将所得物料依次用质量分数2%的稀盐酸洗、质量分数5%碳酸钾的洗、水洗和饱和食盐水洗,将所得洗涤物料分出有机层,将所得有机层物料使用无水硫酸钠干燥,过滤,浓缩至干,采用甲醇重结晶,得到中间体9。
[0155]
在本发明中,所述中间体9具有式i所示结构:
[0156]
得到中间体9后,本发明将所述中间体9、溴化钠、碘化钾和第八溶剂混合,进行溴代反应,得到中间体10。在本发明中,所述中间体9制备中间体10的过程优选与采用中间体3制备中间体5的过程相同(包括原料用量比例和条件),在此不再赘述。
[0157]
在本发明中,所述中间体10具有式g所示结构:
[0158]
得到中间体10后,本发明将所述中间体10与硫代醋酸钾混合,进行取代反应,得到中间体11。在本发明中,利用中间体10制备中间体11的过程优选与采用中间体5制备中间体6的过程相同(包括原料用量比例和条件),在此不再赘述。
[0159]
在本发明中,所述中间体11具有式k所示结构:
[0160]
得到中间体11后,本发明将所述中间体11与碱液混合,进行水解,得到具有式iv所示结构构型的美罗培南侧链光学异构体,记为2s4r-m。在本发明中,所述中间体11水解的过程与中间体6水解的过程相同(包括原料比例和条件),在此不再赘述。
[0161]
在本发明中,所述2s4r-m的合成过程如下式所示:
[0162][0163]
本发明提供了上述技术方案所述美罗培南侧链光学异构体或上述技术方案所述制备方法制备得到的美罗培南侧链光学异构体作为标准品在检测美罗培南侧链产品杂质中的应用。
[0164]
本发明提供了一种美罗培南侧链杂质的检测方法,包括以下步骤:
[0165]
采用高效液相色谱法,以美罗培南侧链光学异构体作为杂质标准品,进行液相色谱检测,得到标准液相色谱谱图;所述美罗培南侧链光学异构体为上述技术方案所述美罗培南侧链光学异构体或上述技术方案所述制备方法制备得到的美罗培南侧链光学异构体;
[0166]
以美罗培南侧链样品作为待测样品,进行液相色谱检测,得到待测样品液相色谱
谱图;
[0167]
将所述待测样品液相色谱谱图与标准液相色谱谱图进行对比,按照面积归一化法计算待测样品中杂质含量。
[0168]
本发明采用高效液相色谱法,以美罗培南侧链光学异构体作为杂质标准品,进行液相色谱检测,得到标准液相色谱谱图。在本发明中,所述液相色谱检测所用杂质标准品溶液的配制方法优选为将美罗培南侧链光学异构体与稀释液混合,得到杂质标准品溶液。在本发明中,所述稀释液优选为正己烷与乙醇的混合液,所述正己烷与乙醇的体积比优选为1:1。
[0169]
本发明对所述液相色谱检测所用高效液相色谱仪没有特殊的限定,符合gb/t 16631-2008高效液相色谱法通则的要求,具备紫外分光光度检测器的仪器即可,具体如shimazu lc-20at。
[0170]
在本发明中,所述液相色谱检测的色谱条件优选为:色谱柱:chiralpakic,250mm
×
4.6mm,5μm;流动相:正己烷、乙醇、甲醇和三氟乙酸的混合物,所述正己烷、乙醇、甲醇和三氟乙酸的体积比优选为(500~700): (200~400):(50~200):(1~10),更优选为600:300:100:1;流速:1.0~1.5 ml/min;检测波长:270nm;进样量:20~25μl;柱温:30~60℃,更优选为40℃;采集时间:20~50min,更优选为35min。
[0171]
以美罗培南侧链样品作为待测样品,进行液相色谱检测,得到待测样品液相色谱谱图。在本发明中,所述美罗培南侧链样品为市售美罗培南侧链或者生产美罗培南过程中所产生的关键中间体美罗培南侧链。
[0172]
在本发明中,所述液相色谱检测所用待测样品溶液的配制过程优选为将美罗培南侧链样品与稀释液混合,得到待测样品溶液。在本发明中,所述稀释液与上述杂质标准品溶液所用稀释液相同,在此不再赘述。
[0173]
在本发明中,所述待测样品的液相色谱检测条件优选与上述杂质标准品的液相色谱检测条件完全相同,在此不再赘述。
[0174]
得到标准液相色谱谱图和待测样品液相色谱谱图后,本发明将所述待测样品液相色谱谱图与标准液相色谱谱图进行对比,按照面积归一化法计算待测样品中杂质含量。在本发明中,进行所述对比时,满足所述美罗培南侧链样品与美罗培南侧链光学异构体之间的分离度≥1.4。
[0175]
本发明对所述采用面积归一化法计算待测样品中杂质含量的具体过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0176]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0177]
实施例1
[0178]
1)(2r,4r)-4-羟基-1-(对硝基苄氧羰基)吡咯烷-2-羧酸(中间体1)的合成:
[0179]
向反应瓶中加入氢氧化钠(66.9g,1.68mol)和水600g,搅拌溶解后,再加入顺式-d-羟脯氨酸(100g,0.76mol)。搅拌全溶后降温到0℃,滴加50%氯甲酸对硝基苄酯的二氯甲烷溶液(370g,0.86mol),温度不超过5℃;滴完后在0-5℃反应3小时,反应结束后,静置分
层,水相用100ml二氯甲烷提杂。分出水相,用10%盐酸调ph至2-3,然后用乙酸乙酯(200ml
×
3)萃取三次,合并有机相,用饱和盐水洗,无水硫酸钠干燥,过滤,浓缩至干,用乙醇重结晶得白色固体221.0g(中间体1),收率93.4%;核磁数据为:1h nmr(d2o,400mhz):δ2.03(m,1h),2.26(m,1h),3.39-3.53(m,2h), 4.28-4.38(m,2h),4.99-5.15(m,2h),7.37-7.44(m,2h),8.07(m,2h).
[0180]
2)(2r,4r)-2-二甲基氨基甲酰基-4-羟基-1-(对硝基苄氧羰基)吡咯烷(中间体2)的合成:
[0181]
氮气保护下,向反应瓶中加入中间体1(40g,0.13mol)、三乙胺(19.6g, 0.19mol)和二氯甲烷300ml,搅拌全溶,降温至-10℃,滴加氯甲酸异丙酯 (19.0g,0.15mol),加完后搅拌2h,滴加二甲胺水溶液(二甲胺0.18mol),温度不超过0℃,滴完后于0℃反应3小时;反应结束后,缓慢加入200ml 水,搅拌5分钟后静置分层,有机相依次用2%稀盐酸100ml洗,5%碳酸钾100ml洗,水洗和饱和食盐水洗,分出有机层,无水硫酸钠干燥,过滤,浓缩至干,用甲醇重结晶得淡黄色固体粉末35.9g(中间体2),收率82.6%;核磁数据为:1h nmr(cdcl3,400mhz):δ2.04(s,1h),2.05-2.32(m,2h), 2.88-3.13(m,6h),3.59-4.15(m,2h),4.57(m,1h),4.86(m,1h),5.02-5.33(m, 2h),7.42-7.51(m,2h),8.19(m,2h).
[0182]
3)(2r,4r)-2-二甲基氨基甲酰基-4-甲磺酰氧基-1-(对硝基苄氧羰基)吡咯烷(中间体3)的合成:
[0183]
氮气保护下,向四口瓶中加入中间体2(47g,0.14mol)、三乙胺(18.8g, 0.19mol)和二氯甲烷200ml,搅拌全溶,降温至-10℃,滴加甲磺酰氯(17.4g, 0.15mol),温度不超过0℃,滴完后于0℃反应2小时;反应结束后,缓慢加入200ml水,搅拌10分钟后静置分层,有机相依次用2%稀盐酸100ml 洗,5%碳酸钾100ml洗,水洗和饱和食盐水洗,分出有机层,无水硫酸钠干燥,过滤,浓缩至干,用甲醇重结晶得黄色固体粉末55.3g(中间体3),收率95.5%;核磁数据为:1h nmr(cdcl3,400mhz):δ2.27-2.37(m,1h), 2.51-2.71(m,1h),2.91-3.16(m,9h),3.87-3.97(m,2h),4.84-4.93(m,1h), 5.05-5.337(m,3h),7.442-7.52(m,2h),8.22(m,2h).
[0184]
4)(2r,4s)-2-二甲基氨基甲酰基-4-乙酰巯基-1-(对硝基苄氧羰基)吡咯烷(中间体4)的合成:
[0185]
氮气保护下,向反应瓶中加入中间体3(5.5g,0.013mol)、硫代醋酸钾 (3g,0.026mol)和n,n-二甲基甲酰胺50ml,升温至55℃反应3h。反应结束后,降温至室温,加入200ml水,搅拌10分钟,然后用乙酸乙酯提取三次(50ml
×
3),合并有机层,用水洗3次,饱和食盐水洗3次,分出有机层,无水硫酸钠干燥,过滤,浓缩至干得黄色油状物5.0g(中间体4),收率96%,未经纯化直接用于下一步;
[0186]
5)2r4s-m的合成:
[0187]
氮气保护下,向反应瓶中加入中间体4(5g,0.013mol)和二氯甲烷50ml,降温至0~5℃,滴加氢氧化钾(1.7g,0.03mol)的甲醇溶液25ml,温度不超过10℃,滴完后保温反应1.5h,反应结束后,加入70ml水,降温至0℃,用10%盐酸调ph至7,温度不超过5℃,静置分液,有机层用水洗两次 (50ml
×
3)后再用饱和食盐水洗,分出有机层,无水硫酸钠干燥,过滤,浓缩得黄色油状物,柱层析(乙酸乙酯:石油醚=体积比2:1)纯化,得淡黄色固体3.6g,即2r4s-m,收率80.6%。
[0188]
对实施例1制备的终产物进行核磁和质谱表征,结果见图1和图2;所得核磁数据为:1h nmr(cdcl3,400mhz):δ1.72(m,1h),2.14(m,1h),2.28(m, 1h),2.90-3.09(m,6h),3.39(m,1h),3.65(m,1h),4.08(m,1h),4.83(m,1h), 5.06-5.33(m,2h),7.40-7.50(m,2h),8.19(m,2h).质谱数据为:lcms,m/z: calcd for(m+h)+,354.4;found,354.1。以上数据表明,2r4s-m的化学结构正确。
[0189]
实施例2
[0190]
1)(2r,4s)-2-二甲基氨基甲酰基-4-溴-1-(对硝基苄氧羰基)吡咯烷(中间体5)的合成:
[0191]
氮气保护下,向反应瓶中加入实施例1制备的中间体3(20g,0.048mol)、溴化钠(24.8g,0.241mol)、碘化钾(0.2g,0.001mol)和n,n-二甲基甲酰胺 100ml,升温至100℃反应2小时,反应结束后,降温至室温,加入500ml 水,搅拌10分钟,然后用乙酸乙酯提取三次(100ml
×
3),合并有机层,用水洗3次,饱和食盐水洗3次,分出有机层,无水硫酸钠干燥,过滤,浓缩至干,得到棕色油状物,柱层析(乙酸乙酯:石油醚=1:1)纯化,得黄色油状物12g(中间体5),收率62.3%,核磁数据为1h nmr(cdcl3,400mhz):δ 2.10-2.29(m,1h),2.75-2.90(m,1h),2.92-3.07(m,6h),3.58-3.74(m,1h), 4.10-4.29(m,2h),4.60-4.73(m,1h),5.01-5.31(m,2h),7.420-7.50(m,2h), 8.18(m,2h).
[0192]
6)(2r,4r)-2-二甲基氨基甲酰基-4-乙酰巯基-1-(对硝基苄氧羰基)吡咯烷 (中间体6)的合成:
[0193]
向反应瓶中加入中间体5(5.5g,0.013mol)、硫代醋酸钾(3g,0.026mol) 和n,n-二甲基甲酰胺50ml,升温至55℃反应3h;反应结束后,降温至室温,加入200ml水,搅拌10分钟,然后用乙酸乙酯提取三次(50ml
×
3),合并有机层,用水洗3次,饱和食盐水洗3次,分出有机层,无水硫酸钠干燥,过滤,浓缩至干得黄色油状物5.26g(中间体6),收率96.8%,未经纯化直接用于下一步;
[0194]
7)2r4r-m的合成:
[0195]
与实施例1中中间体4的水解过程相同,区别在于:以中间体6(2.2g, 0.006mol)为起始原料进行水解,制备得到淡黄色固体1.7g(即2r4r-m),收率77.3%。
[0196]
对实施例2制备的终产物进行核磁和质谱表征,结果见图3和图4;所得核磁数据为:1h nmr(cdcl3,400mhz):δ1.88(m,2h),2.74(m,1h), 2.91-3.08(m,6h),3.26(m,1h),3.44(m,1h),4.07(m,1h),4.68(m,1h), 5.01-5.31(m,2h),7.40-7.50(m,2h),8.19(m,2h).质谱数据为:lcms,m/z: calcd for(m+h)+,354.4;found,354.1。以上数据表明,2r4r-m的化学结构正确。
[0197]
实施例3
[0198]
1)(2s,4r)-4-羟基-1-(对硝基苄氧羰基)吡咯烷-2-羧酸(中间体7)的合成:
[0199]
向反应瓶中加入氢氧化钠(66.9g,1.68mol)和水600g,搅拌溶解后,再加入反式-l-羟脯氨酸(100g,0.76mol),搅拌全溶后降温到0℃,滴加50%氯甲酸对硝基苄酯的二氯甲烷溶液(370g,0.86mol),温度不超过5℃;滴完后在0~5℃反应3h,反应结束后,静置分层,水相用100ml二氯甲烷提杂;分出水相,用10%盐酸调ph至2~3,然后用乙酸乙酯(200ml
×
3)萃取三次,合并有机相,用饱和盐水洗,无水硫酸钠干燥,过滤,浓缩至干,用乙醇重结晶,得白色固体219.2g(中间体7),收率92.6%。1hnmr(d2o,400mhz): δ2.02(m,1h),2.25(m,
1h),3.44(m,1h),3.52(m,1h),4.28-4.38(m,2h), 5.00-5.16(m,2h),7.37-7.44(m,2h),8.08(m,2h).
[0200]
2)(2s,4r)-2-二甲基氨基甲酰基-4-甲磺酰氧基-1-(对硝基苄氧羰基)吡咯烷(中间体9)的合成:
[0201]
氮气保护下,向反应瓶中加入中间体7(30g,0.10mol)、三乙胺(27.9g, 0.28mol)和二氯甲烷500ml,降温至-10℃,滴加氯甲酸异丙酯(13.1g, 0.11mol),温度不超过0℃,滴完后于0℃反应2小时,降温至-10℃,滴加甲磺酰氯(15.0g,0.13mol),温度不超过0℃,滴完后于0℃反应2h,得到含有中间体8的产物,向所得含有中间体8的产物中滴加质量浓度为40%的二甲胺水溶液(14.2g,0.13mol),温度不超过10℃,滴完后保温反应3小时,反应结束后,依次用2%稀盐酸300ml洗,5%碳酸钾300ml洗,水洗和饱和食盐水洗,分出有机层,无水硫酸钠干燥,过滤,浓缩至干,用甲醇重结晶得黄色固体粉末29.8g(中间体9),收率74.2%。1h nmr(cdcl3, 400mhz):δ2.34(m,1h),2.51-2.70(m,1h),2.91-3.16(m,9h),3.94(m,2h), 4.84-4.93(m,1h),5.05-5.37(m,3h),7.44-7.52(m,2h),8.22(m,2h).
[0202]
3)(2s,4s)-2-二甲基氨基甲酰基-4-溴-1-(对硝基苄氧羰基)吡咯烷(中间体10)的合成:
[0203]
与实施例2中采用中间体3制备中间体5的过程相同,区别在于:以中间体9(15g,0.036mol)为起始原料,制备得到黄色油状物9.8g(中间体10),收率67.8%;核磁数据为:1h nmr(cdcl3,400mhz):δ2.43-2.57(m,2h), 2.89-3.14(m,6h),3.93-4.11(m,2h),4.60(m,1h),4.96(m,1h),5.05-5.35(m,2h) 7.42-7.49(m,2h),8.19(m,2h).
[0204]
4)(2s,4r)-2-二甲基氨基甲酰基-4-乙酰巯基-1-(对硝基苄氧羰基)吡咯烷(中间体11)的合成:
[0205]
与实施例1中中间体3制备中间体4的过程相同,区别在于:以中间体 10(11.9g,0.03mol)为起始原料,制备得到黄色油状物10.3g(中间体11),收率87.6%,未经纯化直接用于下一步;
[0206]
5)2s4r-m的合成:
[0207]
与实施例1中中间体4的水解过程相同,区别在于:以中间体11(5.4g, 0.014mol)为起始原料,制备得到淡黄色固体3.7g,收率76.7%。
[0208]
对实施例3制备的终产物进行核磁和质谱表征,结果见图5和图6;所得核磁数据为:1hnmr(cdcl3,400mhz):δ1.73(m,1h),2.14(m,1h),2.28(m, 1h),2.91-3.09(m,6h),3.39(m,1h),3.64(m,1h),4.08(m,1h),4.83(m,1h), 5.05-5.32(m,2h),7.40-7.50(m,2h),8.19(m,2h);质谱数据为:lcms,m/z: calcd for(m+h)+,354.4;found,354.1。以上数据表明,2s4r-m的化学结构正确。
[0209]
应用例
[0210]
本应用例所用稀释液为正己烷:乙醇=1:1(v/v);所用高效液相色谱仪: shimazu lc-20at;
[0211]
标准品溶液配制:准确称取25mg美罗培南侧链样品于50ml容量瓶中,加入15ml稀释液溶解后再加入稀释液稀释至刻度。
[0212]
待测品溶液配制:分别准确称取25mg美罗培南侧链的三个光学异构体于50ml容量瓶中,加入稀释液溶解并稀释至刻度,配置三个待测品溶液。
[0213]
待测样品溶液配置:准确称取美罗培南侧链与三个光学异构体的混合样品25mg于50ml容量瓶中,加入稀释液溶解并稀释至刻度。
[0214]
按照色谱条件运行空白溶液(稀释液),记录色谱图,在无干扰峰前提下,将所述标准品溶液注入高效液相色谱仪中,进行液相色谱分析,记录色谱图及样品峰面积;
[0215]
色谱条件:
[0216]
色谱柱:chiralpak ic,250mm
×
4.6mm,5μm;
[0217]
流动相:正己烷:乙醇:甲醇:三氟乙酸=600:300:100:1(v/v/v/v);
[0218]
流速:1.0ml/min;
[0219]
检测波长:270nm;
[0220]
进样量:20μl;
[0221]
柱温:40℃;
[0222]
采集时间:35min;
[0223]
分别将标准品溶液、美罗培南侧链的三个光学异构体的待测品溶液注入液相色谱仪中,按照上述色谱条件进行液相色谱分析,记录色谱图及样品峰面积;
[0224]
按照面积归一化法计算美罗培南侧链样品中美罗培南侧链异构体的含量,其中,美罗培南侧链样品与美罗培南侧链异构体之间的分离度不小于1.4,所得谱图分别见图7~10;美罗培南侧链及其三个光学异构体在该检测方法下的保留时间如表1所示。
[0225]
表1美罗培南侧链及其三个光学异构体的液相保留时间
[0226][0227]
由图7~10和表1可知,美罗培南侧链三个光学异构体的手性纯度均可达到99%以上,有望作为标准样品使用。
[0228]
按照上述过程,将所述待测样品溶液注入液相色谱仪中,按照上述色谱条件进行液相色谱分析,记录色谱图及样品峰面积,所得谱图见图11,所得保留时间见表2。
[0229]
表2美罗培南侧链及三个光学异构体混合样的液相保留时间
[0230][0231]
由图11和表2可看出,混合物出现的保留时间与纯净物相比,仅出现略微的偏移,说明该检测方法可将美罗培南侧链及三个光学异构体进行有效的分离和鉴定。
[0232]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人
员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1