一种制备吲哚布芬的方法与流程
1.本发明涉及一种化合物的制备方法,具体涉及吲哚布芬的制备方法。
背景技术:
2.随着社会的进步,人们生活水平的不断提高,膳食结构的改善及劳动强度的降低,社会老龄化的加剧等因素,心脑血管疾病的高危人群也在不断增加。而大多数心脑血管疾病斗鱼血栓形成和血栓栓塞有着密切的关系,导致血栓形成的因素有很多,如血小板聚集、血流淤滞、凝血因子的激活促使凝血酶的形成等。因此,寻找高效低毒的抗血小板聚集药物,能有效的减少心脑血管疾病的发病率和死亡率。
3.吲哚布芬(indobufen),化学名2-[4-(1-氧代-2-异吲哚啉基)苯基]丁酸,是由美国辉瑞公司研发的一种外消旋体混合物,由意大利farmfaliacarlo erba,s.p.a首先研制成功,并于1984年8月在意大利(ich成员国)首先上市。吲哚布芬作为新一代非类固醇类强效抗血小板聚集药物,它能选择的作用于循环的血小板,阻断血栓形成,抑制血小板因子释放而发挥抗血小板聚集作用,这种抑制是可逆的,不改变血浆参数,无损血小板功能,并使变异常的血小板功能恢复正常。它可使外周血管病变患者及间歇性跛行患者的微循环参数和行走距离明显改善,在冠状动脉分流术及股动脉分流术后预防在阻塞方面,与阿司匹林加潘生丁效果相同;在血液透析时它能显著减少透析膜上的血小板沉积物。与同类药物相比,吲哚布芬抑制血小板因子,抗血小板聚集效果是水杨酸的2~5倍,较之有轻微持续时间更短的出血时间。
[0004]
高学民等在《抗凝药吲哚布芬的合成》(中国医药工业杂志,1989,20(11))、发明专利cn106631974a、发明专利cn110229075a、发明专利cn101914055a中公开的吲哚布芬制备路线可总结为图3所示:
[0005]
以2-(4-硝基苯基)丁酸为原料,采用铁粉还原、催化加氢或水合肼还原成2-(4-氨基苯基)丁酸,然后再与邻苯二甲酸酐在酸性条件下环化,最后使用锌粉与氯化氢气体进行还原,经过乙醇重结晶处理可得到吲哚布芬。然而最后锌粉还原反应中无法避免地产生过度还原或还原不彻底的杂质,且难以除去,导致最终产物纯度较低。
技术实现要素:
[0006]
本发明的目的在于,克服上述现有技术的不足,提供了一种吲哚布芬的新的合成方法,不仅工艺路线简单,从工业化角度考虑,有操作简单、原料廉价等诸多优点,用此方法生产的中间体纯度很高,很容易得到满足药典要求的吲哚布芬。
[0007]
为实现上述目的,本发明采用以下技术方案:
[0008]
一种合成吲哚布芬的方法,合成路线如图2所示。
[0009]
进一步的,一种合成吲哚布芬的方法,包括以下具体步骤:
[0010]
1.化合物2的合成
[0011]
将2-(4-硝基苯基)丁酸(化合物1)溶于甲醇中,加入浓硫酸,搅拌下加热回流反
应;
[0012]
较佳的,化合物1与甲醇的用量比例为1︰10(w︰v);浓硫酸与化合物1 的用量比例为0.1︰1(v︰w)。
[0013]
2.化合物3的合成
[0014]
化合物2溶于溶剂a中,加入金属还原剂,在室温下加入酸,反应结束后除去溶剂,加入水和乙酸乙酯,加入碳酸钠固体将ph调至8左右,分液,水层再使用乙酸乙酯萃取两次,合并有机相,干燥,过滤,浓缩滤液,即得化合物3;
[0015]
较佳的,化合物2与溶剂a的用量比例为1︰10(w︰v);酸与化合物2的用量比例为5︰1(v︰w);金属还原剂的用量为3.5eq。
[0016]
较佳的,所述金属还原剂为:还原铁粉、锌粉和铝中的一种或者几种,优选还原铁粉。
[0017]
较佳的,所述酸为:浓盐酸、稀盐酸、乙酸和硫酸中的一种或几种,优选浓盐酸。
[0018]
较佳的,所述溶剂a为:甲醇、乙醇、乙腈、丙酮和四氢呋喃中的一种或者几种,优选甲醇。
[0019]
3.化合物4的合成
[0020]
将化合物3溶于溶剂b中,加入邻羧基苯甲醛和反应试剂,反应结束后,浓缩反应液,加入乙酸乙酯搅拌溶解,加入水萃取三次,保留有机相,干燥,过滤,浓缩滤液,即得类白色固体化合物4;
[0021]
较佳的,化合物3与溶剂b的用量比例为1︰15(w︰v);邻羧基苯甲醛的用量为1.1eq;反应试剂的用量为1.2eq。
[0022]
较佳的,所述溶剂b为:n,n-二甲基甲酰胺、乙腈、二氯甲烷、甲醇、乙醇、四氢呋喃和甲苯中的一种或者几种,优选甲醇。
[0023]
较佳的,所述反应试剂为:四丁基硫酸氢铵、硼氢化钠、硼氢化钾、一氧化碳、三氯化铝、二氢吡啶和甲酸/三乙胺中的一种或者几种,优选硼氢化钠。
[0024]
4.化合物5的合成
[0025]
将化合物4加入甲醇中,在室温下加入1mol/l氢氧化钠溶液,反应结束后除去溶剂,加入水和乙酸乙酯萃取,水相加入活性炭脱色,过滤,滴加稀盐酸,将ph调至5左右,析出类白色固体,过滤,真空干燥即得吲哚布芬粗品。
[0026]
本发明的优点是:
[0027]
1.开发了新路线合成吲哚布芬,整个路线总收率为77.9%,收率稳定,反应过程容易控制,易于工业化生产;
[0028]
2.避免使用有危险性的氢气和氯化氢气体,避免了最后锌粉还原反应中产生的过度还原或还原不彻底的杂质,提高产品纯度;
[0029]
3.优化了反应条件,使用价格较低的邻羧基苯甲醛和硼氢化钠,节约了物料成本;
[0030]
4.使用本发明方法生产的化合物4,外观为类白色固体,且纯度达到99.6%,用此方法生产的吲哚布芬(化合物5),外观为白色固体,单杂控制都小于0.10%,产品总纯度达到99.9%,完全能够满足药典要求。
[0031]
5.采用邻羧基苯甲醛和硼氢化钠与化合物2反应,构成吲哚布芬的骨架结构,是该发明一大技术突破,避免了最后锌粉还原反应中产生的过度还原或还原不彻底的杂质,使
反应的纯化过程更简单,后处理简易,且原料价格低廉,降低了物料成本。
附图说明
[0032]
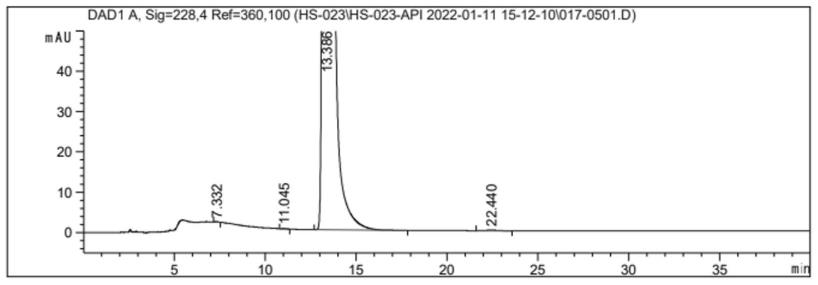
图1是吲哚布芬纯度图。
[0033]
图2是本发明制备吲哚布芬的方法中吲哚布芬的合成路线图。
[0034]
图3是现有技术中公开的吲哚布芬合成路线图。
具体实施方式
[0035]
实施例1
[0036]
1.化合物2的合成
[0037]
将2-(4-硝基苯基)丁酸(50g,1.0eq)溶于甲醇中,加入浓硫酸,搅拌下加热回流反应2h,tlc检测反应结束,反应液可直接投下一步。
[0038]
2.化合物3的合成
[0039]
向上一步的反应液中,加入还原铁粉(46.4g,3.5eq),在室温下滴加浓盐酸,反应3小时,tlc检测反应结束后除去溶剂,加入水和乙酸乙酯,加入碳酸钠固体将ph调至8左右,分液,水层再使用乙酸乙酯萃取两次,合并有机相,干燥,过滤,浓缩滤液,即得化合物3,收率为85.6%。
[0040]
3.化合物4的合成
[0041]
将化合物3(20g,1.0eq),邻羧基苯甲醛(17.1g,1.1eq),硼氢化钠(1.96g, 0.5eq)溶于甲醇中,室温下反应5小时,反应结束后,浓缩反应液,加入乙酸乙酯搅拌溶解,加入水萃取三次,保留有机相,干燥,过滤,浓缩滤液,即得类白色固体化合物4,收率为93.35%。
[0042]
4.化合物5的合成
[0043]
将化合物4(10g,1.0eq)加入甲醇中,在室温下加入1mol/l氢氧化钠溶液,升温至40℃反应3小时,tlc检测反应结束后除去溶剂,加入水和乙酸乙酯萃取两次,水相加入活性炭脱色1小时,过滤,滴加稀盐酸,将ph调至5左右,析出类白色固体,过滤,50℃真空干燥即得吲哚布芬粗品,收率为92.15%。
[0044]
将吲哚布芬粗品(10g,1.0eq)加入乙醇(10v/w)中,升温回流,固体完全溶解后加入活性炭,回流30分钟后,热过滤,滤液冷却至室温后,逐渐溶解后析出白色固体,冰水浴1小时,过滤,少量乙醇冲洗滤饼,滤饼50℃真空干燥,得到吲哚布芬(收率为88.9%,纯度99.97%,如图1)。
[0045]
色谱条件:用十八烷基硅烷键合硅胶为填充剂;以甲醇-水-三乙胺(65:35: 0.5)(磷酸调节ph值至3.0)为流动相;检测波长为228nm。
[0046]
实施例2:
[0047]
1.化合物2的合成
[0048]
将2-(4-硝基苯基)丁酸(50g,1.0eq)溶于甲醇中,加入浓硫酸,搅拌下加热回流反应2h,tlc检测反应结束,反应液可直接投下一步。
[0049]
2.化合物3的合成
[0050]
向上一步的反应液中,加入还原铁粉(60.1g,4.5eq),在室温下滴加浓盐酸,反应3小时,tlc检测反应结束后除去溶剂,加入水和乙酸乙酯,加入碳酸钠固体将ph调至8左右,
分液,水层再使用乙酸乙酯萃取两次,合并有机相,干燥,过滤,浓缩滤液,即得化合物3,收率为85.3%。
[0051]
3.化合物4的合成
[0052]
将化合物3(20g,1.0eq),邻羧基苯甲醛(17.1g,1.1eq),甲酸(7.15g, 1.5eq),三乙胺(15.7g,1.5eq)溶于甲醇中,室温下反应5小时,反应结束后,浓缩反应液,加入乙酸乙酯搅拌溶解,加入水萃取三次,保留有机相,干燥,过滤,浓缩滤液,即得类白色固体化合物4,收率为92.6%。
[0053]
4.化合物5的合成
[0054]
将化合物4(10g,1.0eq)加入甲醇(5v/w)中,在室温下加入1n氢氧化钠溶液,升温至40℃反应3小时,tlc检测反应结束后除去溶剂,加入水和乙酸乙酯萃取两次,水相加入活性炭脱色1小时,过滤,滴加稀盐酸,将ph调至 5左右,析出类白色固体,过滤,50℃真空干燥即得吲哚布芬粗品,收率为92.15%。
[0055]
将吲哚布芬粗品(10g,1.0eq)加入乙醇(10v/w)中,升温回流,固体完全溶解后加入活性炭,回流30分钟后,热过滤,滤液冷却至室温后,逐渐溶解后析出白色固体,冰水浴1小时,过滤,少量乙醇冲洗滤饼,滤饼50℃真空干燥,得到吲哚布芬(纯度99.88%,收率为88.9%,色谱条件同实施例1)
[0056]
实施例3
[0057]
1.化合物2的合成
[0058]
将2-(4-硝基苯基)丁酸(50g,1.0eq)溶于甲醇中,加入浓硫酸,搅拌下加热回流反应2h,tlc检测反应结束,反应液可直接投下一步。
[0059]
2.化合物3的合成
[0060]
向上一步的反应液中,加入锌粉(62.5g,4eq),在室温下滴加浓盐酸,反应3小时,tlc检测反应结束后除去溶剂,加入水和乙酸乙酯,加入碳酸钠固体将ph调至8左右,分液,水层再使用乙酸乙酯萃取两次,合并有机相,干燥,过滤,浓缩滤液,即得化合物3,收率为85.6%。
[0061]
3.化合物4的合成
[0062]
将化合物3(20g,1.0eq),邻羧基苯甲醛(17.1g,1.1eq),二氢吡啶(39.2g, 1.5eq)溶于甲醇中,室温下反应5小时,反应结束后,浓缩反应液,加入乙酸乙酯搅拌溶解,加入水萃取三次,保留有机相,干燥,过滤,浓缩滤液,即得类白色固体化合物4,收率为87.5%。
[0063]
4.化合物5的合成
[0064]
将化合物4(10g,1.0eq)加入甲醇(5v/w)中,在室温下加入1n氢氧化钠溶液(50ml,5v/w),升温至40℃反应3小时,tlc检测反应结束后除去溶剂,加入水和乙酸乙酯萃取两次,水相加入活性炭脱色1小时,过滤,滴加稀盐酸,将ph调至5左右,析出类白色固体,过滤,50℃真空干燥即得吲哚布芬粗品,收率为92.15%。
[0065]
将吲哚布芬粗品(10g,1.0eq)加入乙醇(10v/w)中,升温回流,固体完全溶解后加入活性炭,回流30分钟后,热过滤,滤液冷却至室温后,逐渐溶解后析出白色固体,冰水浴1小时,过滤,少量乙醇冲洗滤饼,滤饼50℃真空干燥,得到吲哚布芬(纯度99.88%,收率为88.9%,色谱条件同实施例1)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1