还原二氧化碳产制碳化合物的方法与流程
本发明是关于一种还原二氧化碳产制碳化合物的方法,尤其是关于一种利用光催化剂还原二氧化碳产制碳化合物的方法。
背景技术:
石化燃料为目前最普遍的动力来源,且在工业发展、交通运输以及农业发展上都占据重要的地位,然而,石化燃料在使用的过程中会排放大量的二氧化碳,造成温室效应、空气污染等环境问题,为了使环境能够永续发展,如何降低二氧化碳的排放量与能源再生为现今重视的议题。
目前降低二氧化碳排放方法系使用高效率的发电系统,但其耗能且需高成本运作不符合经济效益,为了可以节省成本、减少耗能并兼具环境保护,电化学催化与光催化还原二氧化碳为主要的研究技术,其中光催化比起电化学催化最大的优点在于不需要通过电能即能产生催化反应,而是利用太阳光作为能量来源,使用光催化剂进行反应时不会额外制造二氧化碳,因此相关研究学者致力于找寻合适的光催化剂来还原二氧化碳,达到环境保护以及永续能源的发展。
有鉴于此,如何制备出高效能的光催化剂并应用于还原二氧化碳以符合经济效益,遂成相关业者努力的目标。
技术实现要素:
本发明之一目的是在于提供一种还原二氧化碳产制碳化合物的方法,通过合成具有良好光催化性之光催化剂以及复合光催化剂,使其可有效地运用于还原二氧化碳并产制甲烷。
本发明之一实施方式提供一种还原二氧化碳产制碳化合物的方法,其包含提供一光催化剂、提供一还原反应装置、进行一混合步骤、进行一通气步骤以及进行一照光步骤。其中,前述光催化剂包含由下列式(i)、式(ii)、式(iii)、式(iv)或式(v)所示的化合物:
biox式(i)、
mbio2x式(ii)、
liayboc式(iii)、
naayboc式(iv)、
kayboc式(v)。
其中m为铅、钙、锶、钡、铜或铁,x为氟、氯、溴或碘,y为钼、锑、铁、钴、镍、锆、硅、铝或锡,a为0.1至8的正数,b为1至6的正数,c为1至10的正数。前述还原反应装置包含一反应器、一光源以及一第一气体储存装置,光源以及第一气体储存装置皆与反应器连接,且第一气体储存装置系用于储存一二氧化碳气体。前述混合步骤系将光催化剂与一液体溶液于反应器中混合并震荡均匀以形成一混合溶液。前述通气步骤系将二氧化碳气体由第一气体储存装置通入至混合溶液中,使二氧化碳气体饱和溶解于混合溶液中以形成一饱和溶液。前述照光步骤系将饱和溶液于光源下照射,并持续一反应时间,以生成一碳化合物。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中光催化剂可还包含式(i)的化合物与式(ii)的化合物的一复合物、二个式(iii)的化合物的一复合物、二个式(iv)的化合物的一复合物或二个式(v)的化合物的一复合物。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中光催化剂可还包含式(i)的化合物或式(ii)的化合物与一碳纳米材料的一复合物。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中碳纳米材料可为氧化石墨烯(go)或石墨相碳氮化合物(g-c3n4)。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中光源可为可见光、紫外光或太阳光。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中反应时间可为30秒至6小时。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中还原反应装置可还包含一第二气体储存装置,其与反应器连接,且第二气体储存装置系用于储存一氦气气体。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,可还包含一检测步骤,其系利用一检测装置与反应器连接以测量碳化合物的产量。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中检测装置可为气相层析仪。
依据前述实施方式的还原二氧化碳产制碳化合物的方法,其中碳化合物可为甲烷或甲醇。
藉此,本发明的还原二氧化碳产制碳化合物的方法是于可见光的照射下,通过铋基材料及碱金族材料的光催化剂以及复合光催化剂还原二氧化碳制造甲烷。铋基材料及碱金族材料的光催化剂具有优良的光催化性能,可提升光催化效率,以增加甲烷之产率,有效应用于二氧化碳的回收,达到永续发展的目标。
附图说明
为让本发明之上述和其他目的、特征、优点与实施例能更明显易懂,所附图式之说明如下:
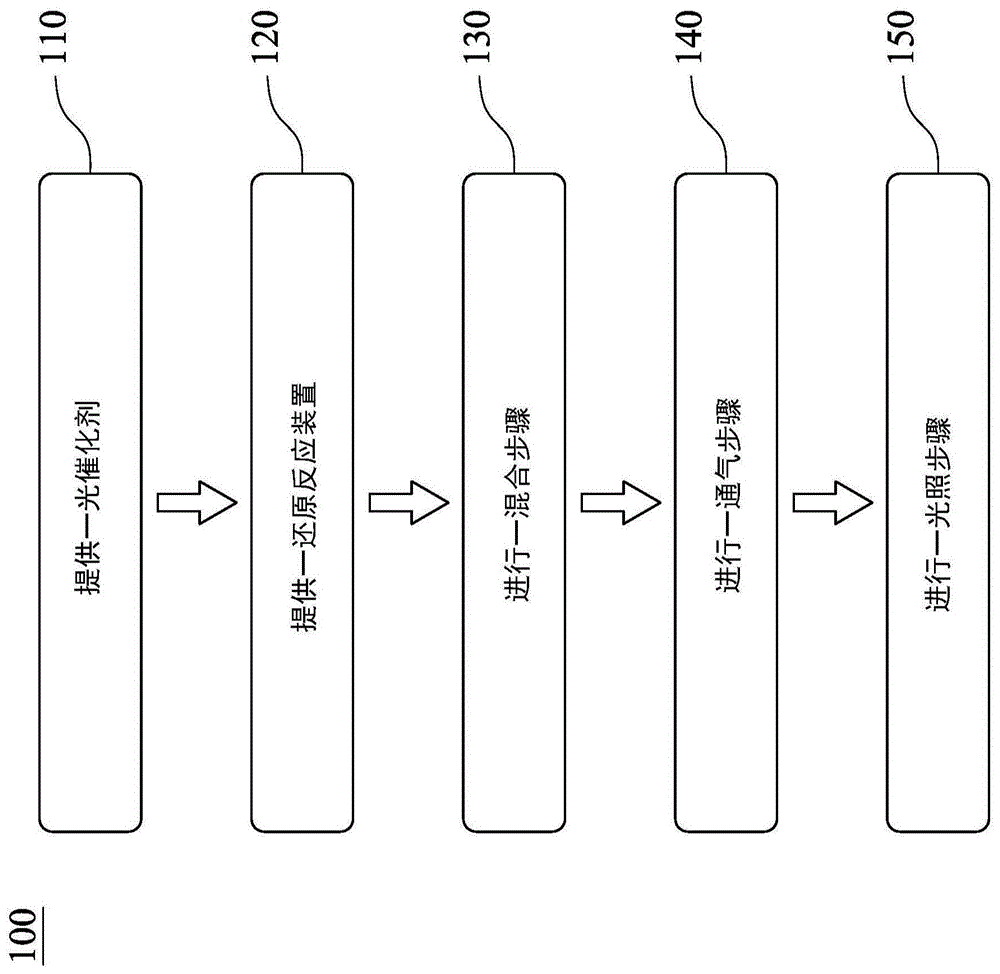
图1系绘示依照本发明之一实施方式的一种还原二氧化碳产制碳化合物的方法的步骤流程图;
图2系绘示依照图1之还原反应装置的示意图;
图3系绘示依照本发明实施例1之光催化剂的xrd绕射分析图;
图4a和图4b系绘示依照本发明实施例1之光催化剂的fesem表面形貌;
图4c系绘示依照本发明实施例1之光催化剂的eds能量分散光谱图;
图5系绘示依照本发明实施例2之光催化剂的xrd绕射分析图;
图6a和图6b系绘示依照本发明实施例2之光催化剂的fesem表面形貌;
图6c系绘示依照本发明实施例2之光催化剂的eds能量分散光谱图;
图7系绘示依照本发明实施例3之光催化剂的xrd绕射分析图;
图8a和图8b系绘示依照本发明实施例3之光催化剂的fesem表面形貌;
图8c系绘示依照本发明实施例3之光催化剂的eds能量分散光谱图;
图9系绘示依照本发明实施例6之光催化剂的xrd绕射分析图;
图10系绘示依照本发明实施例7之光催化剂的xrd绕射分析图;
图11系绘示依照本发明实施例8之光催化剂的xrd绕射分析图;
图12系绘示依照本发明实施例9之光催化剂的xrd绕射分析图;
图13系绘示依照本发明实施例10之光催化剂的xrd绕射分析图;
图14系绘示依照本发明实施例1及实施例11之光催化剂的xrd绕射分析图;
图15系绘示依照本发明实施例11之光催化剂的fesem表面形貌;
图16系绘示依照本发明实施例2及实施例12之光催化剂的xrd绕射分析图;
图17a系绘示依照本发明实施例12之光催化剂的tem明场图;
图17b系绘示依照本发明实施例12之光催化剂的择区电子绕射图(selectedareaelectrondiffraction);
图17c系绘示依照本发明实施例12之光催化剂的hr-tem图;
图17d系绘示依照本发明实施例12之光催化剂的元素分布图;
图17e系绘示依照本发明实施例12之光催化剂的eds能量分散光谱图;
图18系绘示依照本发明实施例3及实施例13之光催化剂的xrd绕射分析图;
图19系绘示依照本发明实施例1及实施例14之光催化剂的xrd绕射分析图;
图20a和图20b系绘示依照本发明实施例14之光催化剂的fesem表面形貌;
图20c系绘示依照本发明实施例14之光催化剂的tem明场图;
图20d系绘示依照本发明实施例14之光催化剂的择区电子绕射图;
图20e系绘示依照本发明实施例14之光催化剂的hr-tem图;
图20f系绘示依照本发明实施例14之光催化剂的元素分布图;
图20g系绘示依照本发明实施例14之光催化剂的eds能量分散光谱图;
图21系绘示依照本发明实施例2及实施例15之光催化剂的xrd绕射分析图;
图22a和图22b系绘示依照本发明实施例15之光催化剂的fesem表面形貌;
图22c系绘示依照本发明实施例15之光催化剂的tem明场图;
图22d系绘示依照本发明实施例15之光催化剂的择区电子绕射图;
图22e系绘示依照本发明实施例15之光催化剂的hr-tem图;
图22f系绘示依照本发明实施例15之光催化剂的元素分布图;
图22g系绘示依照本发明实施例15之光催化剂的eds能量分散光谱图;
图23系绘示依照本发明实施例3及实施例16之光催化剂的xrd绕射分析图;
图24a和图24b系绘示依照本发明实施例16之光催化剂的fesem表面形貌;
图25系绘示依照本发明实施例17之光催化剂的xrd绕射分析图;
图26a系绘示依照本发明实施例17之光催化剂的tem明场图;
图26b系绘示依照本发明实施例17之光催化剂的择区电子绕射图;
图26c系绘示依照本发明实施例17之光催化剂的hr-tem图;
图26d系绘示依照本发明实施例17之光催化剂的元素分布图;
图26e系绘示依照本发明实施例17之光催化剂的eds能量分散光谱图;
图27系绘示依照本发明实施例18之光催化剂的xrd绕射分析图;
图28a系绘示依照本发明实施例18之光催化剂的tem明场图;
图28b系绘示依照本发明实施例18之光催化剂的择区电子绕射图;
图28c系绘示依照本发明实施例18之光催化剂的hr-tem图;
图28d系绘示依照本发明实施例18之光催化剂的eds能量分散光谱图;
图29系绘示依照本发明实施例19之光催化剂的xrd绕射分析图;
图30系绘示依照本发明实施例31之光催化剂的xrd绕射分析图;
图31系绘示依照本发明实施例32之光催化剂的xrd绕射分析图;
图32系绘示依照本发明实施例41之光催化剂的xrd绕射分析图;
图33系绘示依照本发明实施例42之光催化剂的xrd绕射分析图;
图34系绘示依照本发明实施例51之光催化剂的xrd绕射分析图;以及
图35系绘示依照本发明实施例53之光催化剂的xrd绕射分析图。
其中,附图标记说明如下:
100:还原二氧化碳产制碳化合物的方法
110、120、130、140、150:步骤
200:还原反应装置
210:反应器
220:光源
230:第一气体储存装置
231:第一流量控制器
240:第二气体储存装置
241:第二流量控制器
250:管线
260:控制阀
270:搅拌器
280:检测装置
具体实施方式
以下将参照图式说明本发明之实施方式。为明确说明起见,许多实务上的细节将在以下叙述中一并说明。然而,阅读者应了解到,这些实务上的细节不应用以限制本发明。也就是说,在本发明部分实施方式中,这些实务上的细节是非必要的。此外,为简化图式起见,一些习知惯用的结构与元件在图式中将以简单示意的方式绘示;并且重复之元件将可能使用相同的编号表示。
请参照图1以及图2,其中图1绘示依照本发明之一实施方式的一种还原二氧化碳产制碳化合物的方法100的步骤流程图,图2绘示依照图1之还原反应装置200的示意图。还原二氧化碳产制碳化合物的方法100包含步骤110、步骤120、步骤130、步骤140以及步骤150。
步骤110为提供一光催化剂,其中光催化剂包含由下列式(i)、式(ii)、式(iii)、式(iv)或式(v)所示的化合物:
biox式(i)、
mbio2x式(ii)、
liayboc式(iii)、
naayboc式(iv)、
kayboc式(v)。
其中m为铅、钙、锶、钡、铜或铁,x为氟、氯、溴或碘,y为钼、锑、铁、钴、镍、锆、硅、铝或锡,a为0.1至8的正数,b为1至6的正数,c为1至10的正数。详细来说,式(i)的化合物以及式(ii)的化合物皆为铋基材料,铋基材料因其独特的层状结构,有利于电子空穴分离,且在可见光下拥有高活性,使铋基材料具有作为光催化剂材料的潜力。其中,式(i)的化合物系卤氧化铋系列的光催化剂,其结晶构造中含有[bi2o2]2+的层状结构,可与卤素离子(x-)形成双板交错的特殊异相性层状结构,此种特定层状结构可增加其在可见光区域的吸收,并有效促进光电子的分离,然而,卤氧化铋的光催化剂的吸收光区大多在于紫外光区,具有电子-空穴重组率过高的问题,导致其光催化活性常受到限制。为了改善上述问题,进而将金属离子(m2+)掺入[bi2o2]2+的层状结构中,制备出式(ii)的化合物,其系类钙钛矿(perovskite-like)结构的金属卤氧化铋系列的光催化剂,用来修饰式(i)的化合物,提高其光催化活性。
为了增强光催化剂的效率,光催化剂可还包含式(i)的化合物与式(ii)的化合物的一复合物,其可为但不限于pbbio2x/biox或biox/mbio2x。另外,光催化剂可还包含式(i)的化合物或式(ii)的化合物与一碳纳米材料的一复合物,其中碳纳米材料具有优异的化学与物理性质,以及极大的比表面积,使其有良好的电子空穴传导,因此广泛应用于光催化剂复合材料,碳纳米材料可为但不限于碳纳米管、碳纳米纤维、碳纳米球、石墨烯、氧化石墨烯(go)或石墨相碳氮化合物(g-c3n4),较佳地,本发明所使用的碳纳米材料为氧化石墨烯以及石墨相碳氮化合物。
氧化石墨烯具有环氧基、氢氧基等官能基能进行还原反应,使得光催化剂与氧化石墨烯复合时,可将光催化反应产生的光电子导开,以大幅降低电子空穴的重组率,使光催化效率提高,并能够提升光催化剂之比表面积,而式(i)的化合物或式(ii)的化合物与氧化石墨烯的复合光催化剂可为但不限于pbbio2x/go、biox/go、biox/bioy/go或biox/bioy/bioz/go(x、y、z=f、cl、br、i)。石墨相碳氮化合物本身可为可见光催化剂,但是其光催化活性因光生电子空穴重组率高而受限,因此将石墨相碳氮化合物与其他光催化剂复合以制备出异质结构复合光催化剂,可加速分离光生电子空穴,增加光催化效率,而式(i)的化合物或式(ii)的化合物与石墨相碳氮化合物的复合光催化剂可为但不限于pbbio2x/g-c3n4、biox/g-c3n4、biox/bioy/g-c3n4或biox/bioy/bioz/g-c3n4(x、y、z=f、cl、br、i)。
另外,式(iii)、式(iv)以及式(v)的化合物分别为锂、钠、钾等碱金族材料,其可用来作为光催化剂,且可还包含二个式(iii)的化合物的一复合物、二个式(iv)的化合物的一复合物或二个式(v)的化合物的一复合物,其可为但不限于liayboc/liayboc、naayboc/naayboc或kayboc/kayboc。
步骤120为提供一还原反应装置200,如图2所示,还原反应装置200包含一反应器210、一光源220以及一第一气体储存装置230,且光源220以及第一气体储存装置230皆与反应器210连接,其中第一气体储存装置230系用于储存一二氧化碳气体,另外,还原反应装置200可还包含一第二气体储存装置240,其与反应器210连接,且第二气体储存装置240系用于储存一氦气气体。详细来说,第一气体储存装置230与第二气体储存装置240分别连接一第一流量控制器231以及一第二流量控制器241,并经由一管线250连接至反应器210,此外,各气体储存装置以及各流量控制器之间皆设有控制阀260,可控制二氧化碳气体以及氦气气体进入流量控制器,再通入于反应器210中。
步骤130为进行一混合步骤,其系将光催化剂与一液体溶液于反应器210中混合并震荡均匀以形成一混合溶液。如图2所示,混合溶液系将反应器210放置于一搅拌器270上混合震荡所制备而成,所述液体溶液可为碱性氢氧化钠水溶液,其可增加二氧化碳的溶解度。
步骤140为进行一通气步骤,其系将二氧化碳气体由第一气体储存装置230通入至混合溶液中,并维持一小时,确保反应器210内除了二氧化碳外无其他气体残留,使二氧化碳气体饱和溶解于混合溶液中以形成一饱和溶液。
步骤150为进行一照光步骤,其系将饱和溶液于光源220下照射,并持续一反应时间,以生成一碳化合物,其中光源220可为但不限于可见光、紫外光或太阳光,且反应时间可为30秒至6小时。另外,在照光步骤后,可还包含一检测步骤,其系利用一检测装置280与反应器210连接以测量碳化合物的产量,其中检测装置280为气相层析仪。详细的说,在照光反应持续30秒至6小时后,以每30分钟使用检测装置280测量碳化合物以获得各时间点反应之层析图谱数据,以分析碳化合物的产量,而碳化合物可为甲烷或甲醇等有机物。
藉此,本发明之还原二氧化碳产制碳化合物的方法系藉由铋基材料或碱金族材料及其复合物作为光催化剂,并应用于二氧化碳还原制造甲烷等有机物,形成碳循环,达到永续发展的目标。
<实施例>
1.光催化剂的合成
本发明的实施例1至实施例3为pbbio2x(x=cl、br、i)光催化剂,其合成方法系取3mmole的硝酸铋(bi(no3)3·5h2o)溶于10ml的去离子水中搅拌均匀,随后加入1、3、5或15ml的1m硝酸铅(pb(no3)2),并使用氢氧化钠(naoh)水溶液调整ph值,之后再加入1ml的1m卤化钾(kx,x=cl、br、i)水溶液,并剧烈搅拌30分钟,接着放入高压釜中加热至200℃或250℃反应12小时,之后降温至室温后使用去离子水过滤洗涤数次,并置入60℃烘箱烘干12小时,研磨后即可得到实施例1至实施例3。
本发明之实施例4至实施例7为mbio2x(m=ca、sr、ba,x=cl、br、i)光催化剂,其合成方法系取3mmole的硝酸铋(bi(no3)3·5h2o)溶于10ml的1m硝酸(hno3)中搅拌均匀,随后加入1ml的1m卤化钾(kx,x=cl、br、i)搅拌30分钟,并使用氢氧化钠(naoh)水溶液调整ph值,接着放入高压釜中加热至100℃至250℃反应24、48或72小时,之后降温至室温后使用去离子水过滤洗涤数次,并置入60℃烘箱烘干12小时,以产生卤氧化铋。或取0.05mole的硝酸铋溶于25ml的乙醇中配制成a液,取0.05mole的卤化钾溶于25ml之去离子水中配制成b液,将a液与b液混合后剧烈搅拌4小时,以抽气过滤法过滤粉末并烘干,使用玛瑙研钵研磨,也可以产生卤氧化铋。将制备好的卤氧化铋与氢氧化钡(或钙或锶),以摩尔比1:1的比例磨碎混合均匀并放置坩埚中,放入高温炉进行锻烧(调整温度500℃至900℃、时间12至96小时),研磨后即可得到实施例4至实施例7。
本发明的实施例8至实施例10为biox(x=cl、br、i)光催化剂,其合成方法系取3mmole的硝酸铋(bi(no3)3·5h2o)溶于10ml的1m硝酸(hno3)中搅拌均匀,随后加入1ml的1m卤化钾(kx,x=cl、br、i)搅拌30分钟,并使用氢氧化钠(naoh)水溶液调整ph值,接着放入高压釜中加热至100℃至250℃反应24、48或72小时,之后降温至室温后使用去离子水过滤洗涤数次,并置入60℃烘箱烘干12小时。或取0.05mole的硝酸铋溶于25ml之乙醇中配制成a液,取0.05mole的卤化钾溶于25ml之去离子水中配制成b液,将a液与b液混合后剧烈搅拌4小时,以抽气过滤法过滤粉末并烘干,使用玛瑙研钵研磨,也可以产生卤氧化铋。研磨后即可得到实施例8至实施例10。
本发明的实施例11至实施例13为pbbio2x/biox(x=cl、br、i)复合光催化剂,其合成方法系取3mmole的硝酸铋(bi(no3)3·5h2o)溶于10ml的1m硝酸(hno3)中搅拌均匀,随后加入1ml的1m卤化钾(kx,x=cl、br、i)搅拌30分钟,再加入1、3、5或15ml的1m硝酸铅(pb(no3)2),并使用氢氧化钠(naoh)水溶液调整ph值,接着放入高压釜中加热至200℃或250℃反应12小时,之后降温至室温后使用去离子水过滤洗涤数次,并置入60℃烘箱烘干12小时,研磨后即可得到实施例11至实施例13。
本发明的实施例14至实施例16为pbbio2x(x=cl、br、i)/go复合光催化剂,其合成方法系取5mmole的硝酸铋(bi(no3)3·5h2o)溶于5ml的去离子水中搅拌均匀,形成a液,另外,取0.05克的氧化石墨烯(go)溶于10ml的去离子水中搅拌均匀,形成b液,将a液与b液搅拌均匀后加入5或15ml的1m硝酸铅(pb(no3)2),并使用氢氧化钠(naoh)水溶液调整ph值,之后加入1或3ml的5m卤化钾(kx,x=cl、br、i)水溶液,并剧烈搅拌30分钟,接着放入高压釜中加热至200℃或250℃反应12小时,之后降温至室温后使用去离子水过滤洗涤数次,并置入60℃烘箱烘干12小时,研磨后即可得到实施例14至实施例16。
本发明的实施例17至实施例19为pbbio2x(x=cl、br、i)/g-c3n4复合光催化剂,其合成方法系取5mmole的硝酸铋(bi(no3)3·5h2o)溶于5ml的去离子水中搅拌均匀,形成a液,另外,调整g-c3n4与3、5或15ml的1m硝酸铅(pb(no3)2)的比例并溶于10ml的去离子水中搅拌均匀,形成b液,将a液与b液搅拌均匀后使用氢氧化钠(naoh)水溶液调整ph值,之后加入1或3ml的5m卤化钾(kx,x=cl、br、i)水溶液,并剧烈搅拌30分钟,接着放入高压釜中加热至100℃至150℃反应12小时,之后降温至室温后使用去离子水过滤洗涤数次,并置入60℃烘箱烘干12小时,研磨后即可得到实施例17至实施例19。
本发明的实施例20至实施例32系以锂源作为反应物的光催化剂及复合光催化剂,实施例33至实施例42系以钠源作为反应物的光催化剂及复合光催化剂,实施例43至实施例53系以钾源作为反应物的光催化剂及复合光催化剂。上述实施例的合成方法系分别使用碳酸锂、碳酸钠或碳酸钾与三氧化钼、三氧化二锑、氧化铁、氧化钴、氧化镍、二氧化锆、二氧化硅、氧化铝或二氧化锡混合,并以900℃锻烧4小时后取出研磨成粉体,即可得到实施例20至实施例53。
关于本发明的实施例1至实施例53的光催化剂种类如下表一所示:
2.光催化剂的性质分析
2.1卤氧化铋铅光催化剂的性质分析
请参考图3、图4a、图4b以及图4c,其中图3绘示依照本发明实施例1之光催化剂的xrd绕射分析图,图4a和图4b绘示依照本发明实施例1之光催化剂的fesem表面形貌,其中图4a的放大倍率为5000倍,图4b的放大倍率为10000倍,图4c绘示依照本发明实施例1的光催化剂的eds能量分散光谱图。由图3的结果可见,实施例1的光催化剂的加热温度为200℃,其特征峰皆符合pbbio2cl的特征峰(jcpdscardno75-2096),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例1为pbbio2cl光催化剂。另外,由图4a、图4b以及图4c可知,实施例1的光催化剂呈片状结构,且其含有pb、bi、o、cl的化学元素组成,关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表二所示:
请参考图5、图6a、图6b以及图6c,其中图5绘示依照本发明实施例2的光催化剂的xrd绕射分析图,图6a和图6b绘示依照本发明实施例2的光催化剂的fesem表面形貌,其中图6a的放大倍率为5000倍,第64b图的放大倍率为10000倍,图6c绘示依照本发明实施例2的光催化剂的eds能量分散光谱图。由图5的结果可见,实施例2的光催化剂的加热温度为250℃,其特征峰皆符合pbbio2br的特征峰(jcpdscardno38-1008),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例2为pbbio2br光催化剂。另外,由图6a、图6b以及图6c可知,实施例2之光催化剂呈板状结构,且其含有pb、bi、o、br的化学元素组成。关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表三所示:
请参考图7、图8a、图8b以及图8c,其中图7绘示依照本发明实施例3的光催化剂的xrd绕射分析图,图8a和图8b绘示依照本发明实施例3的光催化剂的fesem表面形貌,其中图8a的放大倍率为5000倍,图8b的放大倍率为10000倍,图8c绘示依照本发明实施例3的光催化剂的eds能量分散光谱图。由图7的结果可见,实施例3的光催化剂的加热温度为200℃,其特征峰皆符合pbbio2i的特征峰(jcpdscardno78-0521),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例3为pbbio2i光催化剂。另外,由图8a、图8b以及图8c可知,实施例3之光催化剂呈片状结构,且其含有pb、bi、o、i的化学元素组成。关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表四所示:
2.2卤氧化铋钡光催化剂的性质分析
请参考图9以及图10,其中图9绘示依照本发明实施例6的光催化剂的xrd绕射分析图,图10绘示依照本发明实施例7的光催化剂的xrd绕射分析图。由图9的结果可见,实施例6的光催化剂的加热温度为600℃,加热时间分别为24、48以及72小时,上述特征峰皆符合babio2cl之特征峰(jcpdscardno83-0442),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例6为babio2cl光催化剂。由图10的结果可见,实施例7的光催化剂的加热温度为800℃,加热时间分别为24、48以及72小时,上述特征峰皆符合babio2br的特征峰(jcpdscardno51-0266),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例7为babio2br光催化剂。
2.3卤氧化铋光催化剂的性质分析
请参考图11、图12以及图13,其中图11绘示依照本发明实施例8的光催化剂的xrd绕射分析图,图12绘示依照本发明实施例9的光催化剂的xrd绕射分析图,其中图13绘示依照本发明实施例10之光催化剂的xrd绕射分析图。由图11的结果可见,实施例8的光催化剂的特征峰皆符合biocl的特征峰(jcpdscardno06-0249),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例8为biocl光催化剂。由图12的结果可见,实施例9的光催化剂的特征峰皆符合biobr的特征峰(jcpdscardno78-0348),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例9为biobr光催化剂。由图13的结果可见,实施例10的光催化剂的特征峰皆符合bioi的特征峰(jcpdscardno10-0445),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例10为bioi光催化剂。
2.4卤氧化铋铅/卤氧化铋复合光催化剂的性质分析
请参考图14以及图15,其中图14绘示依照本发明实施例1及实施例11的光催化剂的xrd绕射分析图,图15绘示依照本发明实施例11的光催化剂的fesem表面形貌。由图14的结果可见,实施例11的光催化剂为实施例1的光催化剂与biocl所复合的复合光催化剂,其特征峰皆符合pbbio2cl之特征峰(jcpdscardno75-2096)以及biocl之特征峰(jcpdscardno06-0249),为多晶相化合物,因此,由xrd绕射分析可确认实施例11为pbbio2cl/biocl复合光催化剂。另外,由图15可知,实施例11的光催化剂呈片状结构。
请参考图16、图17a、图17b、图17c、图17d以及图17e,其中图16绘示依照本发明实施例2及实施例12的光催化剂的xrd绕射分析图,图17a绘示依照本发明实施例12的光催化剂的tem明场图,图17b绘示依照本发明实施例12的光催化剂的择区电子绕射图,图17c绘示依照本发明实施例12的光催化剂的hr-tem图,图17d绘示依照本发明实施例12的光催化剂的元素分布图,图17e绘示依照本发明实施例12的光催化剂的eds能量分散光谱图。此外,为使图17d的元素分布清楚明白,实审参考资料中的附件1为图17d的彩色图。
由图16的结果可见,实施例12之光催化剂为实施例2的光催化剂与biobr所复合的复合光催化剂,其特征峰皆符合pbbio2br之特征峰(jcpdscardno38-1008)以及biobr的特征峰(jcpdscardno78-0348),为多晶相化合物,因此,由xrd绕射分析可确认实施例12为pbbio2br/biobr复合光催化剂。另外,由图17a的结果可见,其系由不同大小不同形貌的薄片组成,由图17b的结果可见,其为多晶相型态,与图16的xrd绕射分析的结果相同,由图17c的结果可见,其显示两个不同的晶格条纹,其晶格间距(d-spacing)为0.193nm和0.641nm分别对应于biobr的(113)绕射面和pbbio2br的(002)绕射面,更加确认实施例12为pbbio2br/biobr复合光催化剂,并通过图17d以及图17e可知,实施例12的光催化剂含有pb、bi、o、br的化学元素组成,且皆分布于整个片状上。关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表五所示:
请参考图18,其绘示依照本发明实施例3及实施例13的光催化剂的xrd绕射分析图。由图18的结果可见,实施例13的光催化剂为实施例3的光催化剂与bi5o7i所复合的复合光催化剂,其特征峰皆符合pbbio2i之特征峰(jcpdscardno78-0521)以及bi5o7i的特征峰(jcpdscardno48-0548),为多晶相化合物,因此,由xrd绕射分析可确认实施例13为pbbio2i/bi5o7i复合光催化剂。
2.5卤氧化铋铅/氧化石墨烯复合光催化剂的性质分析
请参考图19、图20a、图20b、图20c、图20d、图20e、图20f以及图20g,其中图19绘示依照本发明实施例1及实施例14的光催化剂的xrd绕射分析图,图20a和图20b绘示依照本发明实施例14的光催化剂的fesem表面形貌,其中图20a的放大倍率为1000倍,图20b的放大倍率为3000倍,图20c绘示依照本发明实施例14的光催化剂的tem明场图,图20d绘示依照本发明实施例14的光催化剂的择区电子绕射图,图20e绘示依照本发明实施例14的光催化剂的hr-tem图,图20f绘示依照本发明实施例14之光催化剂的元素分布图,图20g绘示依照本发明实施例14之光催化剂的eds能量分散光谱图。此外,为使图20f的元素分布清楚明白,实审参考资料中的附件2为图20f的彩色图。
由图19的结果可见,实施例14的光催化剂的加热温度为200℃,其特征峰皆符合pbbio2cl的特征峰(jcpdscardno75-2096),但未观察到go的绕射峰,可能是因为加入go的含量较低,所以go的绕射峰强度相对于pbbio2cl较弱。另外,由图20a以及图20b的结果可见,其由原本实施例1之片状聚集的形貌转变为由纳米薄片聚集形成的花球状,而由图20c的结果可见,颜色较透明的部分为go,颜色较深的为纯相pbbio2cl光催化剂,由图20d的结果可见,其为多晶相(polycrystalline)结构,由图20e的结果可见,其显示晶格条纹及晶格间距为0.1741nm对应于pbbio2cl的(131)绕射面,更加确认实施例14为pbbio2cl/go复合光催化剂,并通过图20f以及图20g可知,实施例14之光催化剂含有pb、bi、o、cl、c的化学元素组成,且皆均匀分布于整个片状上。关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表六所示:
请参考图21、图22a、图22b、图22c、图22d、图22e、图22f以及图22g,其中图21绘示依照本发明实施例2及实施例15的光催化剂的xrd绕射分析图,图22a和图22b绘示依照本发明实施例15的光催化剂的fesem表面形貌,其中图22a的放大倍率为1000倍,图22b的放大倍率为3000倍,图22c绘示依照本发明实施例15的光催化剂的tem明场图,图22d绘示依照本发明实施例15的光催化剂的择区电子绕射图,图22e绘示依照本发明实施例15的光催化剂的hr-tem图,图22f绘示依照本发明实施例15之光催化剂的元素分布图,图22g绘示依照本发明实施例15的光催化剂的eds能量分散光谱图。此外,为使图22f的元素分布清楚明白,实审参考资料中的附件3为图22f的彩色图。
由图21的结果可见,实施例15的光催化剂的加热温度为250℃,其特征峰皆符合pbbio2br的特征峰(jcpdscardno38-1008),但未观察到go的绕射峰,可能是因为加入go的含量较低,所以go的绕射峰强度相对于pbbio2br较弱。另外,由图22a以及图22b的结果可见,其由原本实施例2之板状聚集的形貌转变为由纳米薄片聚集形成的花球状,而由图22c的结果可见,颜色较透明的部分为go,颜色较深的为纯相pbbio2br光催化剂,由图20d的结果可见,其为多晶相结构,由图20e的结果可见,其显示晶格条纹及晶格间距为0.2115nm对应于pbbio2br的(114)绕射面,更加确认实施例15为pbbio2br/go复合光催化剂,并通过图22f以及图22g可知,实施例15之光催化剂含有pb、bi、o、br、c的化学元素组成,且皆均匀分布于整个片状上。关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表七所示:
请参考图23、图24a以及图24b,其中图23绘示依照本发明实施例3及实施例16之光催化剂的xrd绕射分析图,图24a和图24b绘示依照本发明实施例16之光催化剂的fesem表面形貌,其中图24a的放大倍率为1000倍,图24b的放大倍率为3000倍。由图23的结果可见,实施例16之光催化剂的加热温度为200℃,其特征峰皆符合pbbio2i之特征峰(jcpdscardno78-0521),但未观察到go的绕射峰,可能是因为加入go的含量较低,所以go的绕射峰强度相对于pbbio2i较弱。另外,由图24a以及图24b的结果可见,其由原本实施例3的片状聚集的形貌转变为由纳米薄片聚集形成的花球状。
2.6卤氧化铋铅/石墨相碳氮化合物复合光催化剂的性质分析
请参考图25、图26a、图26b、图26c、图26d以及图26e,其中图25绘示依照本发明实施例17的光催化剂的xrd绕射分析图,图26a绘示依照本发明实施例17的光催化剂的tem明场图,图26b绘示依照本发明实施例17的光催化剂的择区电子绕射图,图26c绘示依照本发明实施例17的光催化剂的hr-tem图,图26d绘示依照本发明实施例17的光催化剂的元素分布图,图26e绘示依照本发明实施例17的光催化剂的eds能量分散光谱图。此外,为使图26d的元素分布清楚明白,实审参考资料中的附件4为图26d的彩色图。
由图25的结果可见,实施例17的光催化剂由下到上为100%(纯g-c3n4)、75%、50%、25%、10%、5%、0%(纯pbbio2cl),其特征峰符合pbbio2cl之特征峰(jcpdscardno13-0352),但在低的比例条件下,因为加入g-c3n4的含量较低,所以g-c3n4绕射峰强度相对于pbbio2cl较弱,难以观察到g-c3n4。另外,由图26a的结果可见,在g-c3n4薄片上有深暗区块为pbbio2cl,薄膜且具层状结构为g-c3n4,由图26b的结果可见,其为多晶相结构,由图26c的结果可见,其显示晶格条纹及晶格间距为0.278nm和0.308nm分别对应于pbbio2cl的(020)绕射面和pbbio2cl的(004)绕射面,而g-c3n4则为非晶排列,更加确认实施例17为pbbio2cl/g-c3n4复合光催化剂,并通过图26d以及图26e可知,实施例17的光催化剂含有pb、bi、o、cl、c、n的化学元素组成,且皆均匀分布于多层结构上。关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表八所示:
请参考图27、图28a、图28b、图28c以及图28d,其中图27绘示依照本发明实施例18的光催化剂的xrd绕射分析图,图28a绘示依照本发明实施例18的光催化剂的tem明场图,图28b绘示依照本发明实施例18的光催化剂的择区电子绕射图,图28c绘示依照本发明实施例18的光催化剂的hr-tem图,图28d绘示依照本发明实施例18之光催化剂的eds能量分散光谱图。
由图27的结果可见,实施例18的光催化剂由上到下为100%(纯g-c3n4)、75%、50%、25%、15%、10%、5%、0%(纯pbbio2br),其特征峰符合pbbio2br之特征峰(jcpdscardno38-1008)以及g-c3n4的特征峰(jcpdscardno87-1526)。另外,由图28a的结果可见,在g-c3n4薄片上有深暗区块为pbbio2br,薄膜且具层状结构为g-c3n4,由图28b的结果可见,其为多晶相结构,由图28c的结果可见,其显示晶格条纹及晶格间距(d-spacing)为0.1662nm和0.2053nm对应于pbbio2br的(107)绕射面和g-c3n4的(200)绕射面,更加确认实施例18为pbbio2br/g-c3n4复合光催化剂,并通过图28d可知,实施例18的光催化剂含有pb、bi、o、br、c、n的化学元素组成。关于各元素的重量百分比(weight%)与原子百分比(atom%)如下表九所示:
请参考图29,其绘示依照本发明实施例19的光催化剂的xrd绕射分析图。由图29的结果可见,实施例19的光催化剂由下到上为100%(纯g-c3n4)、75%、50%、25%、10%、5%、0%(纯pbbio2i),其特征峰符合pbbio2i之特征峰(jcpdscardno38-1007),但在低的比例条件下,因为加入g-c3n4的含量较低,所以g-c3n4绕射峰强度相对于pbbio2i较弱,难以观察到g-c3n4。
2.7锂系光催化剂及复合光催化剂之性质分析
关于锂系光催化剂及复合光催化剂的性质分析,以下将以实施例31与实施例32作为描述。请参考图30以及图31,其中图30绘示依照本发明实施例31之光催化剂的xrd绕射分析图,图31绘示依照本发明实施例32之光催化剂的xrd绕射分析图。由图30及图31的结果可见,实施例31之光催化剂的特征峰皆符合li(fe5o8)的特征峰(jcpdscardno82-1436),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例31为li(fe5o8)光催化剂。另外,实施例32的光催化剂为lifeo2与li(fe5o8)所复合的复合光催化剂,其特征峰皆符合lifeo2之特征峰(jcpdscardno74-2284)及li(fe5o8)之特征峰(jcpdscardno38-0259),为多晶相化合物,因此,由xrd绕射分析可确认实施例32为lifeo2/life5o8复合光催化剂。另外,实施例20至实施例30的性质分析方法与实施例31相同,皆能以xrd绕射分析确认其结构,故在此不另赘述。
2.8钠系光催化剂及复合光催化剂的性质分析
关于钠系光催化剂及复合光催化剂的性质分析,以下将以实施例41与实施例42作为描述。请参考图32以及图33,其中图32绘示依照本发明实施例41的光催化剂的xrd绕射分析图,图33绘示依照本发明实施例42的光催化剂的xrd绕射分析图。由图32及图33的结果可见,实施例41的光催化剂的特征峰皆符合nafeo2的特征峰(jcpdscardno76-0600),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例41为nafeo2光催化剂。另外,实施例42的光催化剂为na2feo4与nafeo2所复合的复合光催化剂,其特征峰皆符合na2feo4之特征峰(jcpdscardno01-0835)及nafeo2之特征峰(jcpdscardno74-1351),为多晶相化合物,因此,由xrd绕射分析可确认实施例42为na2feo4/nafeo2复合光催化剂。另外,实施例33至实施例40的性质分析方法与实施例41相同,皆能以xrd绕射分析确认其结构,故在此不另赘述。
2.9钾系光催化剂及复合光催化剂的性质分析
关于钾系光催化剂及复合光催化剂的性质分析,以下将以实施例51与实施例53作为描述。请参考图34以及图35,其中图34绘示依照本发明实施例51的光催化剂的xrd绕射分析图,图35绘示依照本发明实施例53的光催化剂的xrd绕射分析图。由图34及图35的结果可见,实施例51的光催化剂的特征峰皆符合ksbo3之特征峰(jcpdscardno49-0165),且无其他晶相产生,为纯相化合物,因此,由xrd绕射分析可确认实施例51为ksbo3光催化剂。另外,实施例53的光催化剂为实施例51及实施例52的光催化剂所复合的复合光催化剂,其特征峰皆符合ksbo3的特征峰(jcpdscardno49-0165)及k0.51sb2.67o6.26的特征峰(jcpdscardno70-0653),为多晶相化合物,因此,由xrd绕射分析可确认实施例53为ksbo3/k0.51sb2.67o6.26复合光催化剂。另外,实施例43至实施例50以及实施例52的性质分析方法与实施例51相同,皆能以xrd绕射分析确认其结构,故在此不另赘述。
3.还原二氧化碳产制甲烷
本发明系依照图1实施方式的还原二氧化碳产制碳化合物的方法100来进行,详细来说,先将本发明的实施例1至实施例53的光催化剂0.2g加入至300ml的氢氧化钠溶液中震荡均匀形成混合溶液,之后以每分钟50ml的速率通入氦气并维持30分钟后关闭气体使用气相层析仪测量b1作为背景值1,以进行无光照、无二氧化碳的空白测试。接着以每分钟10ml的速率通入二氧化碳并维持60分钟后使用气相层析仪b2作为背景值2,以进行无光照的空白测试。二氧化碳气体不关闭,使其饱和溶解于混合溶液中形成饱和溶液,开启led光源开始照光反应4小时,并且30秒及每30分钟使用气相层析仪测量甲烷产量,以获得各时间点反应之层析图谱数据,反应完成后将光源及气体关闭,将反应后的溶液过滤后在相同参数下使用气相层析仪分析。
本发明之实施例1至实施例10的光催化剂经由照光反应后,皆能还原二氧化碳并产制甲烷,其甲烷随照光时间所产生的浓度、催化剂量、测量时间点以及甲烷产率(μmol/g/h)如下表十所示:
本发明之实施例11至实施例19之复合光催化剂经由照光反应后,皆能还原二氧化碳并产制甲烷,其甲烷随照光时间所产生之浓度、催化剂量、测量时间点以及甲烷产率(μmol/g/h)如下表十一所示:
本发明之实施例20至实施例53之光催化剂及复合光催化剂经由照光反应后,皆能还原二氧化碳并产制甲烷,其甲烷随照光时间所产生之浓度、催化剂量、测量时间点以及甲烷产率(μmol/g/h)如下表十二所示:
综上所述,本发明提供一种还原二氧化碳产制碳化合物的方法,通过合成有光催化效能的铋基材料或碱金族材料及其复合物作为光催化剂,经由照光反应,使光催化剂产生电子空穴,其不仅能还原二氧化碳,达成二氧化碳的减量,同时又可生成具有经济价值的甲烷,以达到永续发展的目标。
虽然本发明已以实施方式揭露如上,然其并非用以限定本发明,任何熟习此技艺者,在不脱离本发明之精神和范围内,当可作各种之更动与润饰,因此本发明之保护范围当视后附之申请专利范围所界定者为准。
- 还没有人留言评论。精彩留言会获得点赞!