1,1’-二吲哚甲烷类衍生物的电化学合成方法与流程
本发明属于有机电合成化学领域,涉及合成1,1’-二吲哚甲烷类衍生物的方法,具体涉及一种以N-取代吲哚及衍生物与四氢呋喃及四氢呋喃衍生物为原料合成1,1’-二吲哚甲烷类衍生物类的方法。
背景技术:
很多具有生物活性的天然产物都含有吲哚类衍生物,尤其是近年来人们从一些陆地和海洋生物体中分离出的1,1’-二吲哚甲烷类化合物表现出更好的生物活性,比如冠状动脉扩张特性、基因毒性、抗菌活性及抗癌活性。因此,引起了不少有机化学工作者的兴趣,进行了广泛的合成方法研究。
目前,主要的合成方法是醛或酮在质子酸或路易斯酸促进下与吲哚或吲哚衍生物的缩合,如2011年,Qu等人用RuCI3做催化剂,苯做溶剂,催化吲哚与醛类反应生成1,1’-二吲哚甲烷类衍生物(Qu,H.E;Xiao,C.;Hu,Q.S.;Wang,N,;Yu,K.H.;Liu,L.X.Molecules 2011,16,3855)。同年,Dashbasi等用In(OTf)3做催化剂,四氢呋喃做溶剂,回流,催化N-取代的吲哚和酰基磷酸盐反应生成1,1’-二吲哚甲烷类衍生物(Dashbasi,T.;Polat Cakir,S.;Abdullah,M.;Demir,A.S.
Tetrahedron 2011.67,3355)等。
此外,2005年,Ma等人用5%Sc(OTf)3做催化剂,乙腈为溶剂,室温下,由α-连烯酮与吲哚分步缩合成1,1’-二吲哚甲烷类衍生物(Org.Lett.,Vol.7,No.22,2005)。2009年,Yu等人用α-不饱和酮二缩醛与吲哚在三氟乙酸催化下合成1,1’-二吲哚烯酮类衍生物(Angew.Chem.2009,121,2973–2977)。也有人用胺类与吲哚缩合,如2011年,Ramachandiran等人用Pb(OAc)2做催化剂,Cu(OAc)2做氧化剂,无溶剂条件下催化吲哚与三乙安反应生成1,1’-二吲哚甲烷类衍生物(Ramachandiran,K.;Muralidharan,D.:Perumal.P.T.Tetrahedron Lett.2011,52,3579)。2009年,Li等人一锅法合成了对称和不对称的1,1’-二吲哚甲烷类衍生物,他们采用FeCI2做催化剂,(t-BuO)2做氧化剂,四氢呋喃做溶剂,氮气保护,80℃下搅拌反应(J.Org.Chem.Vol.74,No.22,2009,8848-8851)。上述举例,从醛、酮类,胺类、醚类描述了1,1’-二吲哚甲烷类衍生物的合成方法。从反应条件来看,使用路易斯酸催化或质子酸催化时,多数情况下都需要当量或者过量,有些情况甚至需要加热或回流;使用金属催化时,有时需要加入过量氧化剂促进金属氧化还原进行循环,或者使用的金属为贵金属,成本较高;当然,也有条件较为简单的,但是又具有底物特殊性,具有一定的局限性。
有机合成化学的发展逐渐朝绿色环保的方向进行,光化学、电化学逐渐表现出优势。以通电的方式取代氧化、还原剂,靠电流的电子作为清洁氧化、还原剂不光不会造成反应试剂残留给分离纯化带来麻烦,而且避免了传统氧化剂、还原剂的使用,避免了环境污染物的生成。
技术实现要素:
本发明在传统有机化学方法合成1,1’-二吲哚甲烷类衍生物的基础上引入电有机合成的绿色观,提供了一种以电子作为清洁氧化剂的方法合成目标产物即1,1’-二吲哚甲烷类衍生物的电化学合成方法。
本发明的合成路线如下所示:
本发明目的通过以下技术方案实现。
1,1’-二吲哚甲烷类衍生物的电化学合成方法,包括如下步骤:
1)将催化剂氯化镧、电解质高氯酸锂、吲哚衍生物加入到电解溶剂中,插入电极,室温下搅拌并通电反应;
2)反应完毕后,萃取、分离纯化,得到产物1,1’-二吲哚甲烷类衍生物。
优选的,步骤1)所述的吲哚衍生物的通式如下所示:
其中,-R1为-H、-F、-Cl、-Br、-I、-NO2、-CN、-OCH3、-CH3或-COOCH3;-R2为-CH3、-CH2CH2CH2CH3或-Bn;-R3为-H、-CH3或-Ph。
优选的,步骤1)所述的电解溶剂为四氢呋喃(THF)或其衍生物和乙腈(MeCN)的混合溶剂。
优选的,所述四氢呋喃或其衍生物和乙腈的体积比为1:1-3:1。进一步优选为2:1。
优选的,步骤1)所述的催化剂为氯化镧(LaCl3),加入量为吲哚衍生物物质的量的10%-30%。
优选的,步骤1)所述的电解质为高氯酸锂(LiClO4),所述电解质在电解溶剂中的摩尔浓度为0.1mmol/ml-0.3mmol/ml。
优选的,步骤1)所述的电极中阴极和阳极距离10mm,阳极为直径0.03mm的铂丝;阴极为10mm×15mm的铂片、10mm×15mm的铜片、10mm×15mm的玻碳电极或直径5mm的碳棒。
优选的,步骤1)所述反应的电流强度为4mA-6mA。
优选的,步骤1)所述的反应的时间为3h-5h。
优选的,步骤2)所得产物为1,1’-二吲哚甲烷类衍生物,结构通式如下:
其中-R1为4、5、6、7-位未取代或取代-F、-Cl、-Br、-I、-NO2、-CN、-OCH3、-CH3、-COOCH3等基团,-R2为1-位取代的-CH3、-CH2CH2CH2CH3、-Bn等基团;-R3为2-位取代的-CH3、-Ph等基团。
以上方法合成的产物1,1’-二吲哚甲烷类衍生物的收率为45%-95%。
与现有技术比较,本发明具有如下优点:
(1)本发明的方法反应物适用范围广,反应选择性好、收率高,适合大规模的工业化生产;
(2)本发明通过电化学手段以清洁氧化剂-电子来氧化,避免了使用化学计量的传统氧化剂,从而避免氧化剂的还原物排放,污染环境。
(3)本发明条件温和,不需要高温,整个操作过程仅需要在传统的搅拌反应装置上通上直流电,简单易行,成本低。
附图说明
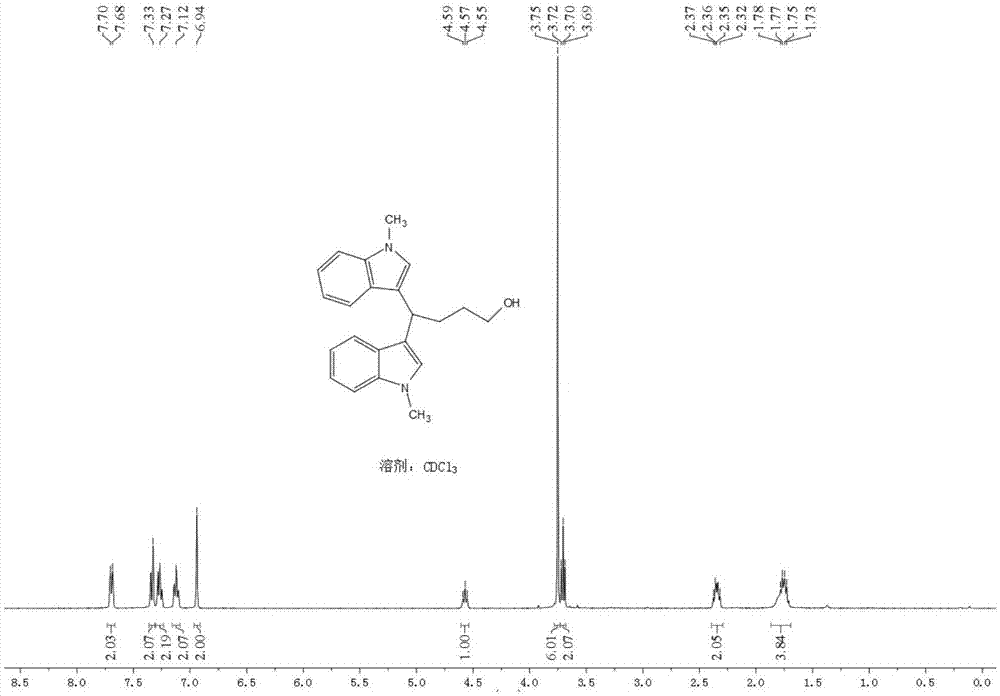
图1为本发明制备的产物1的1H-NMR图谱。
图2为本发明制备的产物1的13C-NMR图谱。
图3为本发明制备的产物2的1H-NMR图谱。
图4为本发明制备的产物2的13C-NMR图谱。
图5为本发明制备的产物3的1H-NMR图谱。
图6为本发明制备的产物3的13C-NMR图谱。
图7为本发明制备的产物4的1H-NMR图谱。
图8为本发明制备的产物4的13C-NMR图谱。
图9为本发明制备的产物5的1H-NMR图谱。
图10为本发明制备的产物5的13C-NMR图谱。
图11为本发明制备的产物6的1H-NMR图谱。
图12为本发明制备的产物6的13C-NMR图谱。
具体实施方式
下面结合实施例及附图对本发明做进一步详细的描述,但本发明的实施方式不限于此。
实施例1
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(0.2mmol/ml)LiCIO4,104.9mg(0.8mmol)N-甲基吲哚;然后加入四氢呋喃3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物1,产率82.4%。
实施例2
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,104.9mg(0.8mmol)N-甲基吲哚;然后加入四氢呋喃3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电2.5mA or 8mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物1产率32.8%(2.5mA)or 45.7%(8mA)。
实施例3
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,104.9mg(0.8mmol)N-甲基吲哚;然后加入四氢呋喃/乙腈(v/v)(1:1or 3:1)混合溶剂5ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物1产率47.5%(1:1)or 62.3%(3:1)。
实施例4
向圆底瓶中依次加入9.6mg(0.04mmol)or 39.0mg(0.16mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,104.9mg(0.8mmol)N-甲基吲哚;然后加入四氢呋喃/乙腈(2:1)混合溶剂5ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物1产率75.1%(5mol%LaCI3)or 71.0%(20mol%LaCI3)。
实施例5
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,53.2mg(在电解溶剂中的浓度为0.1mmol/ml)or 159.6mg(在电解溶剂中的浓度为0.3mmol/ml)LiCIO4,104.9mg(0.8mmol)N-甲基吲哚;然后加入四氢呋喃3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物1,产率68.3%(0.1mmol/ml LiCIO4)or 54.2%(0.3mmol/ml LiCIO4)。
实施例6
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,124.8mg(0.8mmol)N-甲基-5-氰基吲哚;然后加入四氢呋喃3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物2产率92.6%。
实施例7
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,116.1mg(0.8mmol)1,2-二甲基吲哚;然后加入四氢呋喃3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物3产率45.2%。
实施例8
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,151.3mg(0.8mmol)N-甲基-5-甲酸甲酯基吲哚;然后加入四氢呋喃3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物3,产率76.2%。
实施例9
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,116.1mg(0.8mmol)1,5-二甲基吲哚;然后加入四氢呋喃3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物3,产率52.5%。
实施例10
向圆底瓶中依次加入19.5mg(0.08mmol)LaCl3,106.4mg(在电解溶剂中的浓度为0.2mmol/ml)LiCIO4,104.9mg(0.8mmol)N-甲基吲哚;然后加入二氧六环3.3ml,乙腈1.7ml。插入两支电极(铂丝为阳极,铂片为阴极),直流电源供电5mA,搅拌反应,TLC监测,4.5h反应完全。用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和NaCI水溶液(40ml×1)洗,无水Na2SO4干燥,减压蒸干,分离得产物3,产率55.8%。
分析实施例1,4,5,6得到的产物结构,并与Zhiping Li等[J.Org.Chem.2009,74,8848-8851];Khadijeh Ghanbari等[Monatsh Chem.
2014,145,1867-1871]报道的文献对照,结果证实该物质具有产物1,2,3,4结构。
以上实施例得到的产物1(图1、图2)的1H NMR图和13C NMR图如附图所示,鉴定数据如下:
1H NMR(400MHz,CDCl3,ppm)δ7.70(d,J=8.0Hz,2H),7.35(d,J=8.0Hz,2H),7.27(t,J=8.0Hz,2H),7.14(t,J=8.0Hz,2H),6.95(s,2H),4.59(t,J=8.0Hz,1H),3.75(s,6H),3.72(t,J=7.6Hz,2H),2.37(q,J=4Hz,2H),1.75(m,J=8.0Hz,3H).
13C NMR(100MHz,ppm)δ137.4,127.6,126.3,121.4,119.8,118.9,118.6,109.2,63.1,33.7,32.7,32.5,31.6.
以上实施例得到的产物2(图3、图4)的1H NMR图和13C NMR图如附图所示,鉴定数据如下:
1H NMR(400MHz,CDCl3,ppm)δ7.78(s,2H),7.37(d,J=8.0Hz,2H),7.31(d,J=8.0Hz,2H),7.08(s,2H),4.41(t,J=8.0Hz,1H),3.78(s,6H),3.69(t,J=8.0Hz,2H),2.29(q,J=8.0Hz,2H),1.79(s,1H),1.64(q,J=8.0Hz,2H).
13C NMR(100MHz,ppm)δ138.9,128.5,126.8,125.2,124.5,121.0,118.9,110.3,101.6,62.6,33.7,33.1,31.5,31.1
以上实施例得到的产物3(图5、图6)的1H NMR图和13C NMR图如附图所示,鉴定数据如下:
1H NMR(400MHz,CDCl3,ppm)δ7.84(d,J=8.0Hz,2H),7.30(d,J=8.0Hz,2H),7.21(t,J=8.0Hz,2H),7.11(t,J=8.0Hz,2H),4.59(t,J=8.0Hz,1H),3.73(t,J=8.0Hz,2H),3.64(s,6H),2.65(q,J=8.0Hz,2H),2.41(s,6H),1.7(m,J=8.0Hz,3H).
13C NMR(100MHz,ppm)δ136.7,132.9,127.3,120.0,119.6,118.7,114.4,108.6,63.2,35.6,32.3,31.6,29.4,10.9.
以上实施例得到的产物4(图7、图8)的1H NMR图和13C NMR图如附图所示,鉴定数据如下:
1H NMR(400MHz,CDCl3,ppm)δ8.37(s,2H),7.89(d,J=12Hz,2H),7.25(d,J=8.0Hz,2H),6.95(s,2H),4.56(t,J=8.0Hz,2H),3.88(s,6H),3.71(s,6H),3.67(t,J=8.0Hz,2H),2.32(q,J=8.0Hz,2H),1.79(s,1H),1.70(m,J=8.0Hz,2H).
13C NMR(100MHz,ppm)δ168.4,139.8,127.7,126.8,122.9,122.6,120.6,120.2,108.9,62.9,51.8,33.6,32.9,32.4,31.5.
以上实施例得到的产物5(图9、图10)的1H NMR图和13C NMR图如附图所示,鉴定数据如下:
1H NMR(ppm)δ7.43(s,2H),7.18(d,J=8Hz,2H),7.04(d,J=8.0Hz,2H),6.82(s,2H),4.46(t,J=8.0Hz,2H),3.70-3.67(m,8H),2.46(s,6H),2.26(m,2H),2.24(m,2H).
13C NMR(100MHz,ppm)δ135.8,127.7,127.6,126.4,122.9,119.3,118.3,108.8,63.2,33.6,32.7,32.6,31.7,21.6.
以上实施例得到的产物6(图11、图12)的1H NMR图和13C NMR图如附图所示,鉴定数据如下:
1H NMR(400MHz,CDCl3,ppm)δ7.62(d,J=8.0Hz,2H),7.30(d,J=8Hz,2H),7.22(t,J=8.0Hz,2H),7.07(t,J=8Hz,2H),6.90(s,2H),4.88(t,J=8.0Hz,2H),4.15(d,J=8Hz,2H),3.73(s,6H),3.64(m,J=8.0Hz,2H),3.62(m,2H).
13C NMR(100MHz,ppm)δ137.2,127.5,126.9,121.5,119.6,118.7,115.7,109.2,72.0,61.7,34.5,32.7.
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所做的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!