流体分离和检测的制作方法
相关申请
本案要求2013年11月14日(14.11.2013)提交的gb1320146.2的权益和优先权,该案的内容通过引用以其全文结合在此。
本发明涉及用于分析组分混合物(如多肽混合物)的流动方法(如流动扩散方法)和流动装置。
背景技术:
蛋白质-蛋白质相互作用的研究是引起相当大的兴趣的领域。然而,蛋白质相互作用典型地受传统的测量和检测方案所扰乱。用于检查蛋白质-蛋白质相互作用的现有方法包括fret、nmr、epr、spr、esi-ms、尺寸排阻色谱法、以及非变性page。这些方法中的每一种要求相互作用配偶体中的任一个以某种方式被修饰,如通过安装荧光标记物或固定在表面上,或者整个复合体通过基质过筛。这些步骤扰乱了在观察下的瞬态相互作用,具有可能在分离和标记的过程中破坏一些附聚的物种的危险。
流体流(如微流体流)内的组分的分离和检测提出了许多挑战。鉴于最近在用于组分的反应、分离和检测的流体技术方面的增加的兴趣,存在对开发允许组分在连续流动系统中进行分离和分析的方法以及装置的兴趣。
诸位发明人最近已经描述了用于分布组分(包括多组分混合物中的组分)跨过流体装置中的层流的改进的方法(参见pct/gb2013/052757)。组分跨过层流的分布通过荧光光谱法在多个流动时间点进行测量。根据这些测量,有可能识别在流内的不同大小的组分。工作实例示出了使用所描述的方法用于识别aβ(1-42)聚集事件,包括从原始单体物种形成低聚物和细纤维簇。
然而,这项工作必然要求使用具有荧光活性、或设置有荧光标记的组分。在后一种情况下,具有标记物的组分的行为可能受该标记物的影响。诸位发明人的早期工作没有描述来自结合的层流的组分的纯化,也没有建议这可以如何实现。因此,虽然单体和低聚物蛋白质种类被识别,它们不会从流中除去。
流内标记和分离技术是本领域中已知的并且被拉姆齐小组(ramseygroup)很好地描述(例如liu等人分析化学(anal.chem.)2000,72,4608;jacobson等人分析化学(anal.chem.)1994年,66,4127;jacobson等人分析化学(anal.chem.)1994年,66,3472)。例如,该小组已经描述了在流动装置上使用共价和非共价标记的蛋白质的电泳分离(liu等人)。在此,该小组确认在分离之前,特别是在电泳分离实验中标记蛋白质的问题。在流动装置内,该小组建议在分离之后的组分的下游标记,而不是在分离之前的上游标记。电泳技术被用于通过该装置吸引组分。在此,电泳技术基于组分通过毛细管的迁移速度使其暂时分开。以这种方式,具有不同电荷与尺寸之比的组分沿着流体流分布。通过举例,该小组示出了α-乳清蛋白、β-乳球蛋白b和β-乳球蛋白a的分离。标记技术的效率没有进行讨论并且没有建议组分被定量标记。
本发明的诸位发明人现在已经建立了一种用于分离组分(例如呈原状态的蛋白质)并且然后随后在用于检测的优化条件下分析分离的组分的替代流体方法。
技术实现要素:
本发明提供了一种使用流体技术分析组分的方法。该方法采用组分跨过接触流体流(contactingfluidflow)如层状流体流的分布,并分离该分布的一部分用于分析。组分的分布是通过组分从一个流体流扩散或电泳运动进入邻近的流体流如层状流体流可获得的。组分的分布允许组分从流体流内的其他组分中分离。分析可以包括为了便于检测标记组分的步骤。
本发明的方法是一种定量方法,该方法允许组分分离和组分分析独立地进行、并且在对于每一个最佳的条件下的步骤。分离步骤可在天然条件下进行以允许对组分及其环境,包括其与多组分混合物中的其他组分的关系的理解。后续分析可以包括变性和标记步骤以允许分离的组分的精确识别和表征。因此,不必要在其分离之前处理和标记组分。
分离步骤允许感兴趣的组分与跨过流体流的其他组分空间上分离。组分的分离是基于各组分的固有特性,包括大小或电荷。在转向步骤中收集合适地分离的组分,并且在流动条件下、有利地全部在一个流体基质上分析转向流。本发明允许组分在稳态下被分离,从而允许长的曝光时间用于低浓度样品的有效检测。
本发明的流动技术可结合用于制备用于分析的转向的组分的后分离程序。定量标记程序,如在此所述的荧光标记程序,允许从记录的分析信号直接确定组分的浓度。
本发明的方法和装置可用于分析多组分混合物中的组分。此外,这些方法和装置适合于分析在混合物内的组分的缔合和解离。在此描述的技术允许研究多肽组分之间的聚集事件,包括瞬态蛋白质-蛋白质相互作用和通过蛋白和多种可能的粘合剂形成的非强制性蛋白质复合物的行为。因此,相比传统的破坏性检测方案,本发明提供了一种分析聚集和解离事件的替代策略。对缔合和解离事件的分析提供了不间断地定量相对结合动力学的机会。
通过举例,本案表明可以随时间监测胰岛素聚集事件。本发明的方法提供了关于单体和小低聚物在胰岛素聚集反应中的耗尽信息,其中此种信息以前是不可直接取得的。本发明的方法也可用于探测响应于刺激(如ph的变化)的聚集事件,如在此所示的。工作实例还表明扩散方法可用于研究β-乳球蛋白的二聚化。
本发明的方法还允许确定组分的流体动力学半径,包括多相多组分混合物的组分群体的流体动力学半径。
因此,在本发明的一般方面中,提供了一种分析跨过第一和第二接触流体流(如第一和第二层流)分布的组分的方法,该方法包括以下步骤:使该第一流体流的一部分、该第二流体流的一部分、或该第一流体流和该第二流体流的一部分转向,其中该转向的部分包括该组分;并且分析该流体流的转向的部分中的组分。
在本发明的第一方面中,提供了一种用于分析组分的方法,该方法包括以下步骤:
(iii)提供组分跨过第一和第二接触流体流(如层状流体流)的分布;
(iv)使该第一流体流的一部分、该第二流体流的一部分、或该第一流体流和该第二流体流的一部分转向,其中该转向的部分包括该组分;
(v)任选地标记该流体流的转向的部分中的该组分;并且
(vi)分析该流体流的转向的部分中的该组分。
在一个实施例中,该方法包括以下准备步骤:
(i)在一种第一流体流中提供该组分;
(ii)使该流体流与一种第二流体流接触,如此以致产生层流;
以及步骤(iii)是允许该组分加入该第二流体流,从而获得该组分跨过该第一和第二流体流的分布。
在一个实施例中,步骤(ii)包括使该流体流与多个第二流体流接触,如此以致产生这些第二流体流在该第一流体流的任一侧的层流。
在一个实施例中,步骤(iii)包括该组分进入第二流体流的扩散或该组分进入第二流体流的电泳运动。
在一个实施例中,步骤(iv)是使该第二流体流的一部分转向,其中该转向的部分包括该组分。
在一个实施例中,步骤(v)存在。在一个实施例中,步骤(v)是对该组分进行荧光标记。
在一个实施例中,该组分是或包括一种多肽、多核苷酸或多糖。在一个实施例中,该组分是蛋白质。
在一个实施例中,该组分是一种多组分混合物的组分。
本发明还提供了一种适合于在本发明的方法、包括本发明的第一方面的方法中使用的流动装置。
因此,在本发明的另一个方面中,提供了一种用于检测混合物中的组分的流动装置,该装置包括一个用于接触的第一和第二流的分离通道,并且该分离通道与一个下游流分离器流体连通,以及一个在下游且与该流分离器流体连通的检测区,其中该分离通道被适配为允许第一和第二接触流之间的组分的横向运动并且该流分离器被适配为使该第一流体流的一部分、该第二流体流的一部分、或该第一流体流和该第二流体流的一部分从该分离通道转向。
在本发明的另一个方面中,提供了一种用于标记组分的方法,该方法包括以下步骤:
(iii)提供该组分跨过第一和第二接触流体流(如层流)的分布;
(iv)使该第一流体流的至少一部分、该第二流体流的至少一部分、或该第一流体流和该第二流体流的至少一部分转向,其中该转向的部分包括该组分;
(v)标记该流体流的转向的部分中的组分;并且任选地
(vi)分析该流体流的转向的部分中的该组分。
在一个实施例中,该方法包括以下准备步骤:
(i)在一种第一流体流中提供该组分;
(ii)使该流体流与一种第二流体流接触,如此以致产生层流;
以及步骤(iii)是允许该组分加入该第二流体流,从而获得该组分跨过该第一和第二流体流的分布。
在一个实施例中,步骤(iv)是使该第二流体流的至少一部分转向。
附图说明
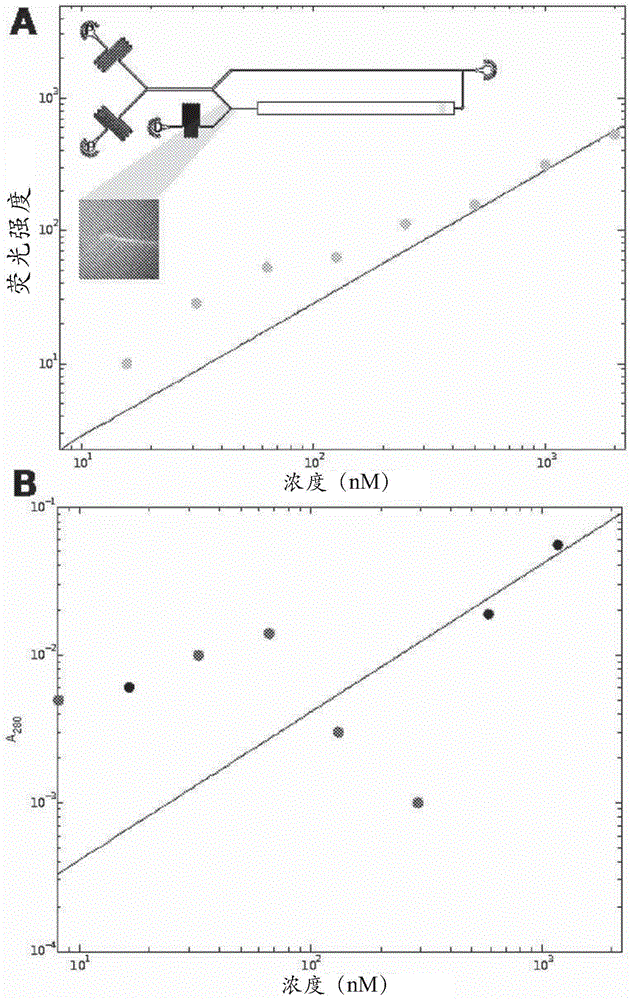
图1示出了(a)根据本发明的一个实施例如在流体装置(插图示出)中测量的随着牛血清白蛋白浓度的变化的荧光强度的变化。bsa在跨过流体流和流分离扩散后被标记。示出了当蛋白质和标记溶液彼此接触时荧光强度的形成。测量并绘制了背景校正的荧光强度随蛋白质浓度变化而变化。观察到约15nm的检测限;以及(b)随牛血清白蛋白浓度的变化在a280的吸光度强度的变化。红点对应于其中样品产生比缓冲液空白更低的吸收的测量。获得约600nm的检测限。
图2示出了对于bsa、溶菌酶和β-乳球蛋白随着有效伯胺浓度变化的荧光强度变化。多种浓度的bsa、溶菌酶和β-乳球蛋白使用在碱性缓冲液中的sds(十二烷基硫酸钠)、opa(邻苯二醛)和bme(β-巯基乙醇)被变性和标记。蛋白质浓度被转换为伯胺浓度,并且示出了伯胺浓度和opa荧光强度之间的线性关系。对于这组变性条件,伯胺浓度和opa荧光强度之间的关系通过低至60nm的蛋白质浓度的线性回归被很好地描述。
图3示出了(a)对于已知浓度的β-乳球蛋白的稀释系列建立的伯胺浓度和opa荧光强度之间的线性关系,其进而被用于确定未知浓度的aβ(1-42)稀释系列的伯胺浓度;以及(b)强度数据到占系列中的稀释的每个β-乳球蛋白样品的蛋白质浓度的转换,由此产生约27μm的一致计算的肽浓度。
图4示出了将10mg/ml的bsa装入扩散装置的两个入口,如在图6和图7(a)中示出的。允许流在分离通道中接触,并且收集层流的一部分。使转向的流与标记流接触,并且此组合流移动穿过混合环路(持续约1.05s),如通过在图6和7(a)中的弯曲通道路径示出的。在样品离开该混合环路的时候,荧光强度是恒定的。代表性迹线重叠,虽然荧光强度沿着该装置的长度保持恒定。这指示标记在约1s混合时间内完成。
图5示出了(a)根据本发明的一个实施例的电泳微流体装置的示意图;(b)示出了与下游流分离器流体连接的分离通道的部分,以及与流分离器的出口连接的标记通道的电泳微流体装置的示意图;以及(c)在ph7下随着跨过分离通道施加的电压的变化的对于偏转的bsa和溶菌酶的归一化荧光强度的变化。bsa和溶菌酶由于它们不同的等电点在ph7下是带相反电荷的。
图6示出了根据本发明的一个实施例的扩散微流体装置的示意图。
图7示出了根据本发明的另一个实施例的扩散微流体装置的示意图。
图8示出了对于具有不同流体动力学半径的颗粒跨过扩散通道的颗粒的模拟数目。在该模拟中,将已知流体动力学半径的颗粒装入像在图6中示出的扩散装置的一个或两个入口中。模拟策略在本文中描述,并且模拟结果对应于随着跨过通道的水平距离变化在扩散通道端部的稳态颗粒强度分布变化。流体动力学半径是比色法表示的。当将颗粒装入两个入口中时,它们的稳态分布不随流体动力学半径(图中的暗水平线)变化,但当将颗粒装入一个入口时它们可预测地随着流体动力学半径变化(演变帽子函数(hatfunction))。
图9示出了对于已经在图6的装置的分离通道中扩散了至少33μm的颗粒流体动力学半径(nm)和所观察的强度比之间的相关性。因此,转向步骤捕获那些具有最小流体动力学半径的组分(并且是因此最迅速地朝向在通道壁的第二流边界扩散)。插入的图是用强度比和流体动力学半径以自然(非对数)标度重新绘制的。
图10示出了(a)根据本发明的一个实施例的扩散微流体装置的示意图,其中该装置使第一和第二流体流的多个部分转向,并且进一步其中每个转向的流的组分在转向后被标记并且随后进行分析;以及(b)对于所观察到的强度比预期的流体动力学半径。
图11示出了对应于图7(c)中显示的ph诱导的胰岛素六聚化数据的原始图像。它们已经根据ph和类型注明。ph2图像示出了当ph2胰岛素接触高离子强度ph10.5标记溶液时,蛋白质如何被破坏并沉积在流体的两个流束之间的层流界面处。虽然最终在下游再次溶解,此蛋白质的沉积可以引起流速的变化。
图12示出了(a)根据本发明的一个实施例的扩散微流体装置的示意图;(b)对于溶剂sds(顶图)和etoh(底图)的(a)的通道在点1、2和3的图像;(c)对于用etoh溶剂标记的bsa、溶菌酶以及β-lac的随着伯胺浓度的变化的荧光强度的曲线图,示出了etoh不溶解,或定量标记所有的蛋白质;以及(d)对于如在喷嘴和(a)中的分区点记录的一系列不同流速随着像素位置变化的荧光强度的曲线图,示出了在扩散通道的不可预测的流动。
图13是图1(a)的扩散微流体装置的放大示意图。
图14是图5(a)的扩散微流体装置的放大示意图。
图15包括在(d)中示意性地示出的微流体装置(具有处理的pdms通道表面)内的通道的明场图像(a)、(b)和(c)以及荧光图像(e)和(f);(a)是在会聚通道的上游端部的接合点的图像;(b)是会聚通道的下游端部的图像;(c)是下游检测区的图像;(e)是会聚通道的下游端部的图像;(f)是下游检测区的图像;以及(g)示出了随时间(s)测量的记录的归一化荧光强度(au),示出了在该通道中提供了稳定的流。
图16示出了如通过(a)动态光散射和(b)如在此描述的基于扩散的流动方法测量的随着β-乳球蛋白浓度(μm)的增加测量的流体动力学半径rh(nm)的变化。该数据示出了相比于本发明的方法动态光散射方法确定流体动力学半径的不可靠性。
图17示出了(a)根据本发明的一个实施例的电泳微流体装置的示意图;以及(b)对于五种不同的蛋白质溶液在电泳微流体装置中随着跨过分离通道施加的电压(v)的变化归一化荧光强度(au)的变化,其中bsa溶液在约4.0v具有最大的荧光强度,β-乳球蛋白溶液在约6.0v具有最大的荧光强度,溶菌酶溶液在约-4.0v具有最大的荧光强度,含有bsa和溶菌酶的溶液在约-7.0v和5.0v具有荧光最大值,以及含有bsa、β-乳球蛋白-和溶菌酶的溶液在约-6.0v、-2.0v、5.0v和6.0v具有荧光最大值。
图18示出了对于处于不同蛋白质浓度的一系列蛋白质样品随时间(s)的记录的荧光强度(au)的变化。这些蛋白质是bsa、lys(溶菌酶)和β-lac(β-乳球蛋白),并且这些蛋白质已经用opa荧光标记混合物标记,如在此所述。
图19示出了对于已经用opa荧光标记混合物处理30分钟的一系列蛋白质样品随着伯胺浓度的变化记录的荧光强度(au)的变化。这些蛋白质是bsa、lys和β-lac。示出了线性拟合。
图20示出了对于一系列从已知浓度的储备样品制备的bsa溶液随着蛋白质浓度(nm)的变化记录的在a280的本体吸收的变化。方形表示其中记录的吸收小于缓冲液空白的数据点。
图21示出了对于在流体装置中越过其iep的牛胰岛素随时间(s)的归一化荧光(au)的变化,其中(a)是根据本发明的实施例的标准pdms流体装置;(b)是已经经受附加的等离子体处理的标准pdms流体装置;以及(c)是已经经受附加的等离子体处理的标准黑色pdms流体装置。
图22(a)示出了对于已知流体动力学半径rh(nm)的参考组分在模拟的扩散梯度中在时刻td的颗粒浓度(au),其中这些组分在具有200μm宽度的模型通道中从第一流体流(虚线左侧)扩散至第二流体流(虚线右侧),并且该组分群体是跨过该扩散通道由侧面观察;(b)示出了对于流体动力学半径rh(nm)的组分观察的强度比,其中这些组分是从第二流体流的一部分转向的,该部分在图22(a)中示出;(c)和(d)是尺寸阶梯实验,对于赖氨酸、胰岛素单体和二聚体的非均匀混合物、β-乳球蛋白二聚体、α-突触核蛋白、bsa、bsa二聚体、以及β-半乳糖苷酶,将通过基于扩散的方法确定的流体动力学半径rh值(“nμ尺寸”;nm)与通过auc和pfg-nmr确定的值(c)和基于组分分子量的预测的最小半径(d)相对比。
图23示出了(a)在定量标记试验中使用的蛋白质的结构的示意图;(b)示出了将蛋白质暴露于opa标记混合物120s后对于它们随着伯胺浓度的变化记录的荧光强度(au)的变化;(c)示出了对于在时刻0用opa标记混合物处理的bsa蛋白质随时间(s)的归一化荧光强度(au)的增加;(d)示出了将蛋白质暴露于opa标记混合物3s后对于它们随着伯胺浓度的变化记录的荧光强度(au)的变化;以及(e)示出了对于bsa蛋白质蛋白浓度(nm)和记录的荧光强度(au)之间的关系,示出了nm蛋白质浓度遵循线性拟合。
图24是根据本发明的一个实施例的扩散微流体装置的示意图,并且表示在图13中示出的装置的改变。
具体实施方式
本发明提供了用于分析流体中的组分(包括多组分混合物中的组分)的方法和装置。本发明的方法包括使第一和第二接触流体的流的一部分转向,并且分析该转向的流的步骤。该流的转向的部分是组分跨过第一和第二流体流的横向分布的一部分。分析步骤有利地是与转向步骤和分离步骤(若存在)一致地执行。
典型地,组分跨过第一和第二流的分布是通过组分从第一流体流扩散传输至第二流体流中获得的。然而,可以使用允许组分从第一流体流至第二流体流的横向运动的其他技术。例如,可以使用电泳技术以获得组分跨过第一和第二流体流的分布。
本发明允许组分以其原状态被分离和转向,例如与也以其原状态的其他组分一起。因此,一种组分或多种组分的横向分布是那些以原状态的组分的扩散或电泳特征的代表。在该组分是多组分混合物的一部分时,在横向分布中各组分的比例是以原状态的那些组分的相对比例的代表。
本发明的方法还适合在分析以非常低的浓度的组分中使用。本发明的方法还使用非常小的样品尺寸,这意味着能够以阿摩尔水平的灵敏度检测组分。可以在四个数量级的浓度范围内测定组分的分子尺寸,同时容许未标记物种的不均匀混合物。
如在此所述,可以使用本发明的方法来分析聚集事件和离解事件。
在转向横向分布的一部分后,没有必要保留以其原状态的组分。后续分析步骤可以在对于识别和定量最佳的条件下进行。
用于分析组分的微型装置是已知的,然而此类装置不被适配为用于分离跨过通道的一种或多种组分。本发明的诸位发明人已经发现,组分跨过流的分布的发展是一种有用的分离策略,该分离策略可以用作分析感兴趣的组分的前导步骤。分离和分析步骤在微流体装置上的结合提供了一种用于研究组分,特别是在复合多组分混合物中存在的那些组分的改进的方法。
一般方法
本发明的第一方面的方法总体上用于分析,如表征或量化,溶液中的组分。
使包含一种或多种组分的第一流体流在分离通道中与第二流体流接触,如此以致产生层流。允许接触的流沿着该分离通道流动并且允许该第一流体流中的组分移动至该第二流体流中,以提供组分跨过该第一和第二流体流的分布。该第一流体流的一部分,该第二流体流的一部分,或该第一和第二流体流的一部分被转向至转向通道中并且然后允许流动至分析通道中用于分析。在分析之前,可以允许在该转向通道中的转向的流接触从试剂通道提供的试剂流。该试剂流提供用于混合物的试剂,并且任选地与组分反应,从而允许该组分的改进的检测和表征。
该分离通道、转向通道以及分析通道和试剂通道(当存在时)是流体装置的部件。该流体装置,特别是该分析通道,被适配为与用于这些组分的检测器一起使用。
在分离、转向和分析步骤过程中,每个流的流速保持在基本上恒定的水平。这些分离、转向和分析步骤可以仅当在每个部分的通道中建立稳定流时进行。
该组分可以是或包含多肽、多核苷酸或多糖。
在一个实施例中,该组分是或包含多肽。在一个实施例中,该组分是或包含蛋白质。
该组分可以是多组分混合物的一部分。因此,该分离步骤可以被用于至少部分地将该组分与其他组分分离。例如,在此描述的技术允许除其他外基于尺寸或电荷与尺寸之比的分离。
在一个实施例中,该多组分混合物含有组分,包括蛋白质,如单体、二聚体和三聚体物种的聚集体,或其他更高级聚集体。因此,在此描述的技术可以用于分离和分析蛋白质-蛋白质相互作用。这在对于蛋白质胰岛素的工作实例中示出。
流体流
本发明提供了用于在流体流中提供的组分的分离和分析的方法。在一个实施例中,对流体流的提及是对液体流的提及。
流体流可以是水性流。水性流可以包括其他溶剂,如dmso、烷基醇以及类似物。
本发明的装置可以被适配为与流体流一起使用,并且可以被适配为与水性液体流一起使用。
在本发明的实施例中,该组分是在第一流体流中初始提供的。该组分优选溶解在该第一流体中。
在一个实施例中,该第一流体允许一种或多种组分保持在其原状态。在该组分是生物分子,如蛋白质时,该流体流可以是合适的缓冲液。因此,可以选择,除其他外,盐含量和ph值以保持该组分以其原状态。
第二流体流可以与该第一流体流相同,除了该第二流体流不含有该组分。
使该第一和第二流体流接触,并且允许该第一流中的组分移动至该第二流中以产生该组分跨过第一和第二流体流的分布。接触流可以是该第一流与该第二流的层流。
在一些实施例中,使标记流与转向的流接触。该标记流典型地是液体流,例如水性流,含有适合于对组分进行标记的试剂。
在一些实施例中,使变性流与转向的流接触。该变性流典型地是液体流,例如水性流,含有适合于使组分变性的试剂。
分离
本发明的方法包括提供组分跨过该第一和第二流体流的分布的步骤。该分布典型地是组分跨过该第一和第二流体流的非均匀分布。
本发明的方法可以包括将组分跨过流体流分布的准备步骤。因此,该组分是在第一流体流中提供的,并且允许该组分与该第二流体流结合,从而提供该组分跨过该第一和第二流体流的横向分布。如在此描述的,该分布可以包括该组分进入该第二流体流的扩散或该组分进入该第二流体流的电泳运动。可以使用其他横向分布技术。
该分布是该组分或包含该组分的多组分混合物的横向分布。
横向分布可以与组分沿流体流的分布不同。例如,本领域已知的是流体方法可被用于基于在流体通道中的物种的泰勒(talyor)分散分离流体流中的组分。例如,us2011/264380描述了用于测定多分散物种的流体动力学半径的方法。将有待分析的物种与单分散标准混合。将所得的混合物添加到沿着一个毛细管流动的载体流体中并且在该混合物排出该毛细管时记录它的泰勒曲线。
如前面提及的,拉姆齐小组已经描述了用于分离蛋白质的电泳方法,然而,这些蛋白质是沿着流体流分离的并且没有组分跨过流的非均匀分布。这可以被认为是暂时的而不是空间分布。如前面指出的,与此相反,本发明允许组分在稳态下被空间分离,从而允许长的暴露时间用于低浓度样品的有效检测。
在此描述的分离方法对于在这些流中使用的溶剂条件的性质很大程度上是不敏感的。因此,可以研究在其天然条件下的生物分子如蛋白质。以此方式组分在分离步骤中的行为是以其原状态的组分的特征。对于分析没有必要包括将组分在外部条件下的行为转换为在自然条件下的预期行为的校准步骤。
在该组分是混合物(多组分混合物)的一部分时,该组分和该混合物的其他组分可以跨过通道干扰,从而提供所有组分跨过该第一和第二流体流的分布曲线。
转向步骤可以在组分到达第二流体流的边界(即,通道壁)之前进行。在该组分是多组分混合物的一部分时,该转向步骤可以在该多组分混合物中的任何组分到达第二流体流的边界之前进行。
分布曲线依赖于用于分布该组分的技术,以及分布所允许的时间。典型地,分布所允许的时间是这样使得在第一流体流中的组分没有到达该第二流体流的边界,如以上所指出的。第一和第二流在分离通道中的流停留时间可被选择为使得在该第一流体流中的组分在所采用的分离条件下没有时间到达边界。
在一个实施例中,该组分的分布可以从该第一流扩散到该第二流。扩散传输速率与该组分的扩散系数d成比例并且与流体动力学半径rh成反比。因此,预期较小的组分以比较大的组分更大的速率跨过该第二流体流扩散。因此,在转向步骤中,靠近在壁处的该第二流体流的边界的该第二流体流的一部分的转向将收集具有较小尺寸的那些组分。靠近具有该第一流体流的层流边界的该第二流体流的一部分的转向将允许收集具有较大尺寸的那些组分。由此得出在层流边界与通道边界之间的该第二流体流的一部分的转向将允许收集中等尺寸的那些组分。
转向的组分的大小将取决于在分离通道中的流分离器的位置。转向的组分的范围将取决于与该第一或第二流体流的总宽度相比转向的部分的相对大小,以及转向的流的部分。
转向步骤可以收集第一流体流的一部分。使用扩散分离技术,预期在该第一流体流中的较小的组分比较大的组分更迅速耗尽,因为较小的组分以更大的速率扩散到该第二流体流中。
在一个实施例中,该组分的分布可以从该第一流电泳运动到该第二流。电泳转移的速率与该组分的电荷与尺寸之比成正比。因此,预期具有大电荷和/或小尺寸的组分相比具有较小电荷和/或尺寸的那些组分具有更大的电泳运动。
在电泳用于分离组分时,该第二流体流典型地在该第一流体流的两侧提供。在电泳过程中带负电荷的物种可以被偏转到第二流体流之一中,而带正电荷的物种被偏转到其他流体流中。
因此,预期具有高电荷与尺寸之比的组分以比具有低电荷与尺寸之比的组分更大的速率移动(转移或偏转)跨过该第二流体流。因此,在转向步骤中,靠近在壁处的该第二流体流的边界的该第二流体流的一部分的转向将收集具有高电荷与尺寸之比的那些组分。靠近具有该第一流体流的层流边界的该第二流体流的一部分的转向将允许收集具有低电荷与尺寸之比的那些组分。由此得出在层流边界与通道边界之间的该第二流体流的一部分的转向将允许收集中等电荷与尺寸之比的那些组分。
转向的组分的电荷与尺寸之比将取决于在跨过分离通道中的流分离器的位置。转向的组分的范围将取决于与该第一或第二流体流的总宽度相比转向的部分的相对大小,以及转向的流的部分。将理解的是,该第二流体流的一部分的转向可收集仅具有负或正电荷的那些物种。
流分离器也可被放置来收集第二流体流之一的一部分,从而收集带正电荷的亦或带负电荷的组分。
以上描述的是用于分布组分跨过第一和第二流体流的扩散和电泳方法。可以使用用于分布组分的替代方法。实例包括例如金属蛋白的等电点测定、超速离心和磁分离。
本发明的方法包括使该第一或第二流的一部分转向,或使该第一和第二流体流的一部分转向的步骤。
该转向步骤不包括使该第一流体流的全部或该第二流体流的全部的步骤。
us2006/0263903描述了一种适合于测量组分从一个流体流扩散至另一个流体流的流动装置。该组分在允许在交叉接合点处接触第二流体流的第一流体流中提供。该第一和第二流体流形成层流,并且允许组分从该第一流体流扩散至该第二流体流。该第一和第二流体流之间的接触时间通常是短的,并且该第一和第二流随后被分离。对该第二流体流进行分析以确定存在的组分的量。
对比于us2006/0263903中描述的方法,本发明没有使该第二或第一流体流的全部转向。相反,本发明的方法使该第一流体流的一部分、该第二流体流的一部分、或该第一和第二流体流的一部分转向。
对流体流的一部分的分析允许用户确定在扩散分布的一部分中的材料的数量并且对其识别。此信息不是us2006/0263903的装置的用户可获得的。
us6,468,761讨论了在用潜在的荧光团的分离步骤之前或之后标记产品。没有讨论使用变性剂以使能定量标记该组分。
kamholz等人描述了一种适合于测量组分从一个流体流扩散至另一个流体流的流动装置。该装置是本领域常见的并且还描述在brody等人和hatch等人中。此类装置用来使组分流体流和空白流体流汇集在一起。该组分和空白流体流形成层流,并且允许组分从该组分流体流扩散至该空白流体流。该组分和空白流体流随后不分离,并且跨过整个空白流体流测量(例如通过荧光)该组分的扩散。
分离步骤可以通过色谱和泰勒分离方法,以及拉姆齐小组的替代电泳技术(毛细管电泳),其中组分沿流体流分离来区分。这样的技术可以被视为及时分离组分。相反,在本案中使用的分离方法在空间上分离组分。
在一个实施例中,允许组分跨过流体流部分地扩散,例如从一个流至另一个流。
在一个实施例中,该第一流体流被提供作为两个层流第二流体流之间的中心流。因此,在该第一流体流中的组分可以被分布到第二流体流的一个或两个中。
在一个实施例中,测量了跨过这些流的一种或多种组分的分布。一种或多种组分的分布可以沿着流体流在多个位置处进行测量。在层流被转向之前进行测量。在使用扩散分布技术时,每个位置代表特定的扩散时间。只有当该组分具有允许它被检测到的固有官能度时可以进行这样的测量。在组分缺乏这样的官能度时,其可以在以后的标记步骤中被提供有官能度。
在本发明中上述步骤不是必要的,因为与该组分的分布有关的信息可从转向的流记录,如在此所述。
转向
本发明的方法包括使该第一和/或第二流体流的一部分转向的步骤。该流体流的转向的部分包含组分,并且组分的分析在该流体流的转向的部分中进行,其是从该第一和第二流体流的其余部分分离的。
该转向步骤取该第一流体流的一部分、或该第二流体流的一部分、或该第一和第二流体流的一部分。在一个实施例中,该转向步骤取该第二流体流的一部分。
该转向步骤并不取该第一流体流的全部或该第二流体流的全部。当提及该第一和第二流体流的一部分的转向时,这是提及该第一流体流的一部分和该第二流体流的一部分的转向。层流的这一部分的转向包括其中该第一流体流和该第二流体流接触的边界。
该转向步骤分离该流体流的一部分用于后续分析。所取的该流体流的部分表示在分离步骤中建立的横向分布曲线的一部分。该转向步骤是该流体流的总宽度的一小部分,或该第一或第二流体流的宽度的一小部分的分离。被转向的该流体流的一小部分没有特别限制并且基于用于分析的组分和,如果存在的话,多组分混合物中的其他组分选择。
该转向步骤指的是对应于该第一流体流和/或第二流体流的一部分的流的一部分的分离。当该第一和第二流体流首先接触时,存在第一和第二流体流之间的明显区别。前者携带组分,而后者不携带组分。在分离通道的下游端,来自该第一流体流的组分移动跨过进入该第二流体流中以产生组分跨过该第一和第二流体流的分布。
在本案中,提及流体流的转向是提及该第一和第二接触流体流的特定截面部分,例如通道中的特定区域。据信通道的该区域是该第一流体流的一部分,如果它对应于在该通道的上游部分的通道中的区域(如接合点),在这里该第一流体流与该第二流体流首先接触。
例如,当该第一和第二流首先接触时,可以在通道的上游部分建立接触流,其中该第一流体流占据通道宽度的一半并且该第二流体流占据通道宽度的剩余的一半。如果流体流的转向的部分取自被该第一流体流最初占据的通道宽度的一半,则该部分可以被称为该第一流体流的转向的部分。在这种情况下,该第一和第二流体流之间的分界简单地是通道中的中心线。
在接触流的下游端的该第一和第二流体的位置可以从用第一流体流保持的组分的分布来确定。例如,在扩散分布中,非常大的组分将具有进入该第二流体流的可忽略的扩散。在下游端非常大的组分将主要保留在该第一流体流中(参见,例如图8,其示出了在根据本发明的一个实施例的分离步骤中存在具有大流体力学半径的组分进入该第二流的最小扩散)。在电泳分离中不带电的组分将具有响应于施加场的可忽略的偏转,并且因此将基本上不会从该第一流体流移出。
在一个实施例中,该转向步骤使至少5%、至少10%、至少15%、至少20%、至少25%、至少30%的该第一流体流,该第二流体流或该第一和第二流体流转向。
在一个实施例中,该转向步骤使最多40%、最多50%、最多60%、最多75%、最多85%的该第一流体流,该第二流体流或该第一和第二流体流转向。
在一个实施例中,该转向步骤使从一定范围的一定量的该第一流体流、该第二流体流或该第一和第二流体流转向,其中该范围的下限和上限值是选自以上给定的最小值和最大值。
在一个实施例中,该转向步骤使该第二流体流的一部分转向。
在一个实施例中,被转向的该第二流体流的部分可以是从该第二流体流与该第一流体流的边界延伸跨过该第二流体流的宽度的最多5%、10%、15%、25%、50%或75%的部分。
在一个实施例中,被转向的该第二流体流的部分可以是从该第二流体流与该通道壁的边界延伸跨过该第二流体流的宽度的最多5%、10%、15%、25%、50%或75%的部分。
在一个实施例中,被转向的该第二流体流的一部分不包括从该第二流体流与该第一流体流的边界延伸的部分或从该第二流体流与该通道壁的边界延伸的部分。因此,该转向的部分是该第二流体流的中间部分。该中间部分可以是该第二流体流的宽度的最多5%、10%、15%、25%、50%或75%。
被引导的流体流的一部分将取决于待检测的组分的特性和分离步骤的性质。
如在以上分离部分中提到的,扩散和电泳分离技术可用于获得一种或多种组分跨过该第一和第二流体流的分布。被转向的流体流的部分可以被选择为分析具有感兴趣的特性的组分,例如特定尺寸或特定电荷与尺寸之比。
本发明的方法可用于收集差别在于感兴趣的特性的组分。所收集的第一或第二流体流的一部分可以改变以转向替代组分。分离技术也可适配为改变在流被转向的点处的组分的分布。例如,扩散分离的扩散时间可以随流量的变化,或者分离通道的长度的变化而改变(如在pct/gb2013/052757中所述的)。在电泳分离中的组分的偏转可以随流速的变化或所施加的场的变化而改变(例如如由herling等人所述的)。
没有必要分离组合流作为流高度(或深度)的一小部分。
在本发明的装置中,流体流可以通过在分离通道的下游端的出口通道的适当放置分离。转向通道可位于适当的侧向位置,以使来自分离通道的第一或第二流体流(或流)的所要求部分的流体转向。
未转向的层流的剩余部分可以被收集,或这些部分可以被分析,如以下进一步详细描述的。
允许在分离通道中的流体流的一部转向入转向通道。在转向通道中的流体与检测区流体连通,如检测区的检测通道,在那里可以分析从分离通道被递送到转向通道的组分。
在本发明的一个实施例中,使多个流体流部分转向。分析层状流体流的至少一个转向的部分。在层流的转向的部分包括第二流体流的一部分时,分析该转向的部分。
每个转向的流是该第一流体流的一部分、该第二流体流的一部分、或该第一流体流和该第二流体流的一部分。转向的部分之一包括组分。在该第一流体流包括多种组分时,多个流体流部分中的每一个可含有组分。
如下面描述在下游随后分析转向的流。
在本发明的一个实施例中,第一和第二流的转向的部分在分析后与该流的其他部分重新组合。因此,在最初的第一和第二流体流中的所有组分可以被收集用于进一步的分析和使用。
用于流体流束的转向的流体装置是在本领域中已知的,但这些装置并不适配为在使含有组分跨过该流的分布的流的一部分转向中使用。
例如,us2002/0186263描述了一种具有多个沿流动通道串联安排的小部分收集器的微流体装置。该装置被设计成使得每个小部分收集器(其简单地是阀)能够引导通道中的所有流进入侧通道。没有建议使该流体流的一部分转向。此外,没有提及组分的分布,如组分的非均匀分布(跨过通道)并且没有提及扩散或电泳技术。
us2010/0032349描述了一种用于从流体流产生液滴的流体装置。没有提及扩散或电泳分离。尽管该文件描述了在该装置的下游端分离所形成的液滴,分离沿着流体流的方向发生,而不是跨过流体流,如由本案的方法所要求的。us2010/0032349并没有提及层状流体流或跨过流体的组分分布,并且没有其中流体流的一部分从另一个转向的步骤的清楚描述。
us2012/0135507主要关注的是在流动装置中使用的基底的性质以及使用磁珠使用该流动装置选择性地捕获细菌。该文件没有描述扩散或电泳分离,并且没有指示存在组分跨过层流的非均匀分布。的确,us2012/0135507似乎没有披露使用层状流动流体。在该文件提及分离时,这似乎只是指磁珠在流体流中被分离,而且没有讨论转向的流体流的比例。
wo2010/004236描述了一种材料分离流动装置。该流动装置包括一个流动屏障,该流动屏障防止材料从主通道进入分支通道。因此,提供了在其内具有组分的流。允许该流沿通道移动并且该通道具有分支。该分支可将该流的一部分,例如含有感兴趣的组分,带到分析装置。
wo2010/004236解释了防止组分进入分支有时是有用的。为了做到这一点,在主流的侧面设置阻挡流,并且跨过分支以防止主流中的组分进入到分支中。因此,在设置第二流时旨在防止组分跨入到分支中。感兴趣的组分可以仅当被诱导这样做时被吸入分支中,例如当在分支侧施加电压以电动引导下游组分的流时。
wo2010/004236用于收集沿流动通道的长度已经分离的组分。wo2010/004236没有描述组分跨过通道的分离和收集。
如前面提到的,us2006/0263903描述了在相交点产生层状流体流的步骤,以及该层流的后续分离。该层流是从含有组分的流和空白流产生的。允许在相交点处的组分扩散到空白流。分离涉及从剩余的含有组分的流使所有空白流(其现在包含一些少量的组分)转向。us2006/0263903没有描述使组分流或空白流的一部分(唯一的)转向的步骤。us2006/0263903显然仅适合用于单一组分使用,并且没有暗示它可能或将适合于分离多组分混合物。
分析
在本发明的方法的步骤(v)中,分析流体流的转向的部分(包含组分)。
该分析步骤可以包括制备流体流的转向的部分,包括制备该组分用于分析的准备步骤。
在一些实施例中,本发明的方法包括允许该组分与该第二流体结合的步骤,其中该组分是处于原状态。在这种形式下该组分可能不适合用于分析。因此,本发明的方法可以包括制备用于分析的组分的步骤,其可以包括或涉及该组分的变性。有利的是,一种或多种组分的分布可以在天然条件下发生,并且后续分析步骤可以在用于组分的最佳表征的替代条件下进行。
典型地,该组分是由光谱法包括uv/可见光和荧光光谱,并且优选通过荧光光谱法进行分析。荧光光谱法因为提供高信噪比是特别有吸引力的。
在一个实施例中,该转向的流与试剂流接触,并且允许该试剂流内的一种或多种试剂与该转向的流中的组分混合,任选地反应。在适当的混合和反应后,进行分析。该组分可以在该流体流内进行分析。该试剂可以是标记物或可在反应时产生可检测的标记物。
在本发明的一个实施例中,该组分在分离后,例如在转向后被标记。标记过程是用于组分的分析的检测步骤的一部分。
添加标记物可能是检测所分离的组分所必要的。例如,该组分可能不具有允许其通过光谱法检测的合适的或足够的官能度。例如,在组分没有或具有很少发色团时,在分析前用一个或多个发色团标记组分可能是有益的。
在一个实施例中,该组分具有一个或多个发色团标记物,例如荧光团标记物(在分离后)。
在一个实施例中,该标记物是潜在标记物。潜在标记物是只有当它与该组分相缔合时是光谱活性(如荧光活性)的标记物。否则,该标记物是非光谱活性的。因此,潜在标记物只有当它与该组分相缔合时是可检测的,并且没有与该组分形成缔合的标记物保持非光谱活性。由此得出该组分的检测被简化,因为没有必要从流体流除去未反应的标记物,或降低标记物对所记录的光谱信号的贡献。
例如,并且如在此所述的,与组分的反应可除去淬灭其荧光的在标记物上存在的基团,该反应从而除去淬灭。在另一个实例中,标记物(例如荧光团基团)在标记反应过程中形成,例如通过形成延伸的共轭体系。
在一个实施例中,该标记物共价键合到该组分。因此,该标记步骤包括在该标记物和该组分之间形成一个或多个共价键。该共价键可以与组分上的氨基、羟基或硫醇基团形成。在该组分是多肽,例如蛋白质时,该共价键可以与氨基酸残基侧链官能度形成。
在其他实施例中,可以使用非共价标记物,其可以是对该组分特定的或非特定的。用于组分的非共价标记物的实例由拉姆齐小组描述(参见,例如,liu等人,分析化学(anal.chem.)2000,72,4068)。
在一个实施例中,该标记物与该组分的氨基官能度,如伯氨基官能度(-nh2)反应。在该组分是或包括多肽,例如蛋白质时,该标记物可以与多肽的赖氨酸残基反应。也可使用与羟基(-oh)、羧基(-cooh)和硫醇(-sh)官能度反应的标记物。在该组分是或包括多肽,例如蛋白质时,该标记物可以与例如多肽的丝氨酸或苏氨酸、天冬氨酸或谷氨酸、或半胱氨酸残基反应。
在一个实施例中,该标记物衍生自邻苯二醛或含邻苯二醛的化合物。此类对于标记氨基官能度是特别有用的,并且对于标记多肽是特别有用的,如在此所述。
本发明的诸位发明人已经发现,邻苯二醛(opa)可以方便地在本发明的流动方法中作为潜在共价标记物使用。opa可以与该组分的一个或多个氨基反应以形成可检测的荧光标记。opa优选地在含有硫醇的试剂(如烷基硫醇,如β-巯基乙醇(bme))的存在下与组分的氨基反应。
在一个实施例中,该标记反应是基本上定量反应。因此,在一个实施例中,基本上所有的转向组分被标记。此外,在组分包含多个能够与该标记反应的基团时,基本上所有的那些基团与该标记反应。因此,所记录的光谱信号可用于直接定量该流中的组分。此外,高度的标记(即所有标记的组分和/或组分具有多个标记)总体上改进了流体流中的组分的检测。这在其中该组分以非常低的浓度存在的流动条件下是特别重要的。
该标记反应应该适合于在流动系统中使用。因此,重要的是该标记反应在相对短的时间范围内发生,因为在装置中的流体的停留时间不长。本发明的诸位发明人已经发现,opa标记物与组分如蛋白质迅速反应,并且因此适合于在此处所描述的流动方法中使用。
在一个实施例中,标记反应时间为最多5s、最多2s、最多1.5s或最多1s。标记反应时间可以是指标记至少50摩尔%、至少80摩尔%或至少90摩尔%的组分(优选90摩尔%)所花费的时间。在一个实施例中,标记反应时间可以是指反应半衰期。
本发明的诸位发明人已经发现,某些标记物在本发明的方法中使用的流动条件下可能是不稳定的。因此,该标记物可能随时间降解,这在分析步骤过程中具有降低检测到的信号强度的影响。因此,存在所记录的组分浓度小于样品内的实际组分浓度的风险。
例如,已知的是从邻苯二醛(opa)与氨基(例如存在于多肽氨基酸残基中的氨基)的反应形成的荧光团缺乏高化学稳定性(参见jacobs等人;daito等人和nakamura等人)。
本案的方法允许标记的组分在标记步骤完成后很快进行分析。当标记的组分的信号强度达到最大值时,该标记步骤可被视为完成。用于定量或识别组分的合适的分析测量可在信号强度达到最大值的时间,或之后不久进行。
在本发明的方法中,诸位发明人已经发现潜在标记物(如邻苯二醛(opa))迅速与组分反应以产生标记的组分。该标记步骤可以在少至三秒内完成,如通过在标记反应过程中将信号强度(例如荧光强度)增加至最大强度判断的。由此得出该组分的分析发生在从标记反应的开始大约三秒,或之后很快。这样的分析使用在此所述的流体系统是完全可行的。
在一个实施例中,本发明的方法包括在分析步骤(vi)之前的标记步骤(v),并且该分析步骤在该标记步骤开始后不久进行。例如,该分析步骤可在该标记步骤开始最多1秒、最多2秒、最多3秒、最多5秒、最多10秒、最多20秒、或最多30秒内进行。
该标记步骤的开始可以是指当首先允许在流体流中的组分与标记试剂,例如在流体接合点处接触时的时间点。
在一些实施例中,没有必要对组分进行标记,因为该组分可能固有地具有使用上述光谱法(如荧光光谱)可检测的官能度。例如,当组分具有荧光活性基团时,这些可用于该组分的荧光检测。
是或包括多肽的组分可以具有氨基酸色氨酸、酪氨酸和苯丙氨酸,其侧链具有荧光活性。然而,这些残基的存在可能不足以允许该组分的检测。例如,酪氨酸和苯丙氨酸荧光活性非常弱,并且因此难以检测。在多肽内存在少量色氨酸、酪氨酸和苯丙氨酸的氨基酸残基时,荧光信号可能是弱的。在这些情况下,可能优选的是提供具有更大的荧光活性的荧光标记物。opa衍生的标记物是可以使用的标记物的一个实例。
当本发明的方法包括标记该转向的组分的步骤时,转向的流与包括标记物的流体流(标记流体流)任选地与用于标记反应的相关试剂一起接触。转向的流和标记物流体流在流分离器的接合点下游汇集在一起。
允许该组分和标记物在流体流内混合,从而标记该组分。可以允许标记物流和转向的流沿混合通道流动以确保在装置内用于标记的足够的时间,例如,以允许在光谱分析之前用于标记的足够的时间。
在一个一般实施例中,该组分的二级、三级和/或四级结构,如二级或三级结构,优选的三级结构,在分离步骤之后、分析之前改变。
使用在此所述的标记方法,本发明的诸位发明人已经发现没有必要破坏组分的二级结构,并且足以改变三级和/或四级结构(当存在时),以允许适当的标记。
在一个实施例中,该组分在分析之前被变性。
该变性步骤旨在提供可能有助于该组分的标记和/或检测的组分上和内的官能团。例如,当该组分是多肽,例如蛋白质时,该变性步骤可以露出氨基、羟基和硫醇官能用于与标记物反应。
该组分的变性可以通过将变性试剂加入到流体流中来实现。例如,当该组分是多肽时,sds可用作变性试剂。
该变性步骤不限于使用变性试剂并且环境改变(如温度)可用来实现变性。
该组分可以在标记前被变性。可以进行变性和标记步骤的分离以最小化组分的沉淀,其可以在组合的变性和标记步骤过程中发生。
在该变性步骤利用变性试剂时,该变性试剂可以在与该转向的流接触的流体流(变性流)中提供。转向的流和变性流体流在流分离器的接合点下游汇集在一起。允许该组分和变性试剂在流体流内混合,从而变性该组分。可以允许变性流和转向的流沿混合通道流动以确保在装置内用于变性的足够的时间,例如,以允许在与标记流(在使用时)接触之前或在光谱分析之前用于变性的足够的时间。
在该方法还包括标记组分的步骤时,该标记步骤在变性步骤的下游进行。
可替代地,该组分可以在一个组合步骤中被变性并且标记。在存在组分的沉淀的小风险时,可以使用组合的变性和标记步骤。因此,在一个实施例中,标记物流体流另外包括变性试剂。如在此所示的,允许转向的流和标记流(含有变性剂)接触的接合点可以适配为处理变性问题。因此,流体通道在接合点处的表面可以是这样使得可使用流体中的排斥组分(例如亲水性表面),以防止疏水性组分粘附在通道表面。
在转向的流与标记物流或变性流接触时,优选的是允许流的内含物和与其接触的流的内含物快速混合。快速混合是确保该组分的快速标记或变性。这应该与使该第一流体流和该第二流体流接触的步骤进行对比,其中没有必要或希望快速分布组分跨过第一和第二流二者。例如,在扩散分离步骤中,在分离通道中早期建立组分的均匀分布是不希望的,因为这将不允许组分被分离。对于扩散分离,有必要跨过第一和第二流体流建立非均匀分布曲线。
流动装置
本发明提供了一种适配为在本发明的方法中使用的流动装置。该流动装置允许第一和第二流体流接触并形成层流。该流动装置被适配为使该第一流体流的一部分、该第二流体流的一部分、或该第一流体流和该第二流体流的一部分转向进入下游转向通道。该转向通道与分析通道流体连通并且由此得出来自转向通道的流被提供到该分析通道用于分析。任选地,允许来自该转向通道的流体流与试剂流体流接触,该试剂流体流是从上游试剂通道提供的。
本发明的流动装置可以是集成装置,诸如单片装置,具有集成的通道网络。因此,该装置不具有死体积并且谱带增宽是有限的。
该流动装置使用小流体通道、特别是微流体通道,并且因此可以分析非常小的样品体积。因此,在小于一微升体积的流体中提供的组分可以通过在此所述的方法进行分析。另外,流体流动技术还可以用于通过适当增加测量次数来分析非常稀的样品。
如在此所述的,本发明的第二方面的流动装置包括用于第一和第二流的分离通道,并且该分离通道与下游流分离器流体连通,以及在下游且与该流分离器流体连通的检测区,其中该分离通道被适配为允许组分在第一和第二流之间的的横向运动并且该流分离器被适配为使该第一流体流的一部分、该第二流体流的一部分、或该第一流体流和该第二流体流的一部分从该分离通道转向。
该分离通道,该转向通道和该检测通道的截面典型地是在微米范围内,并且在本发明的第一方面的方法中使用的流体装置可以因此被称为微流体装置。
本发明还提供在此所述的微流体装置。
使用微流体通道容纳该第一流体和第二流体流确保这些流在低雷诺数下发生。在此处所描述的扩散分离步骤下,对流和扩散是系统内的质量传递的唯一有关机制。因此,这允许对给定尺寸的每种组分进行准确的数值计算,如在此进一步详细描述的。在使用电泳方法用于分离时,对流和电泳是系统内的质量传递的唯一有关机制。
分离通道具有允许在两个(或三个)流束内生成并保持一个层流的适合尺寸。两个流束的层流意指这些流是并排地并且是稳定的。因此,典型地不存在其中这些流体再循环的区域并且湍流是最少的。典型地,通过小通道如微型通道提供此类条件。
选择该装置中的通道的总体尺寸以提供合理的移动速度和分析时间。还可以选择该装置的尺寸以减小持续一个足够的分析运行所需要的流体的量。
在组分跨过流体流的扩散中使用的装置(诸如在分散测量中使用的)是在本领域中众所周知的,并且例如由kamholz等人(生物物理杂志(biophysicaljournal)80(4):1967-1972,2001)描述。
在组分跨过流体流的电泳中使用的装置是在本领域中众所周知的,并且例如由herling等人(应用物理快报(appliedphysicsletters)102,184102-4(2013))描述。因此,该分离通道可以沿着通道长度配备有电极用于偏转(分布)带电组分跨过通道。这是与由拉姆齐小组描述的装置可区分的,其中电极被放置在通道端部,以便沿着通道长度分布组分。
该分离通道是具有允许产生稳定的流体流并且用于实现组分跨过流的充分分离的适合的尺寸的通道。
该分离通道是其中该第一流体流与该第二流体流接触的区域。
在此提及分离通道是提及具有基本上矩形的截面的通道。因此,该分离通道可以是由基本上平的基底(具有基本上垂直于该基底延伸的多个壁)和任选地顶盖形成的。典型地,该基底和这些壁被形成为一个硅酮衬底。该盖可以是一个玻璃盖,例如标准玻璃载片或硼硅酸盐晶片。
典型地,该装置内的其他通道,诸如流分离器,也是基本上矩形的。
分离通道是与用于供应第一流体的一个或多个储罐流体连通的。分离通道是与用于供应第二流体的一个或多个储罐流体连通的。
典型地该流动装置包括一个第一供应通道和一个第二供应通道,这些通道与下游分离通道流体连通。该第一供应通道是用于容纳该第一流体流并且该第二供应通道是用于提供该第二流体流。该第一和第二供应通道在接合点处与下游分离通道相遇,该接合点被适配为以层流容纳第一和第二流体流。这些通道提供储罐与分离通道之间的流体连通。
在一个实施例中,该分离通道包括一个第一大截面通道和在下游并与该大截面通道流体连通的一个第二小截面通道。
本发明的诸位发明人已经发现,在其中该第一和第二流体首先接触的接合点处使用大截面通道最小化了流体滞留。此类通道在pct/gb2013/052757中描述。
这些流体的流动是沿着分离通道的纵轴。一种或多种组分从第一流进入第二流的运动,如一种或多种组分的扩散,是横向于流动的纵轴,跨过通道的宽度。
本发明的流动装置可以结合诸位发明人的早期工作的流动装置,如pct/gb2013/052757中描述的,其内容以其全文通过引用结合在此。
该流动装置包括在分离通道下游并且与其流体连通的流分离器。该流分离器是位于跨过分离通道的一部分的通道以收集层流的一部分,并且尤其是收集该第一流体流的一部分、该第二流体流的一部分或该第一和第二流体流的一部分。该通道的位置和宽度取决于待收集的层流的一部分以及待收集的流的比例进行选择。
该流分离器从该分离通道转向该流的一部分。该流分离器提供转向的流到下游检测区,并且与该下游检测区流体连通。
该检测区包括用于容纳来自上游流分离器的流体流的检测流体通道。该检测区可以包括用于分析容纳在该检测流体通道中的组分的分析装置。
在一个实施例中,该检测流体通道与一个或多个上游流供应通道连通,这些流体通道是在该流分离器的下游。流供应通道是用于供应标记物和变性试剂到该检测流体通道。供应通道的每一个可以与用于容纳相关试剂如标记物和变性试剂的上游储罐连通。
如在此所述的,标记物和变性试剂可以在一个流体流中一起提供。因此,单个供应通道可在该检测通道的上游提供。该供应通道在接合点处接触该检测通道。
如在此所述的,标记物和变性试剂可以在分开的流体流中提供。因此,第一供应通道可以被提供用于递送变性试剂进入该检测通道。第二供应通道可以被提供用于递送标记物进入该检测通道。该第一和第二供应通道分别在第一和第二接合点处与该检测通道接触。该第一接合点位于该第二接合点的上游。
在允许转向的流与标记物流和/或变性流在检测通道中混合时,该检测通道可设有一个混合区以确保在转向的流中的组分与标记物和/或变性试剂的充分混合。该混合区可以简单地指的是为流体提供足够的流动停留时间以允许该组分的混合和反应的检测通道的延长。该混合区可以具有非直线路径以增强混合。使用此类通道结构是本领域技术人员众所周知的。
该分析装置没有特别的限制并且包括那些适合于与流动装置,以及特别是微流体装置使用的装置。可以设置多个分析装置以确定该组分的不同的物理和化学特性。这些分析装置可以顺序地或并联地安排。
该分析装置可以结合考虑的组分标记物,或考虑的组分的固有光谱特性进行选择。
在一个实施例中,该分析装置是荧光计。
在一个实施例中,该分析装置是一个干质量测量装置,诸如石英晶体微量天平。本发明的方法和装置可以与gb1320127.2的干质量方法和装置一起使用。
在一个实施例中,该装置包括用于从分析区收集流输出的储罐。
在一个实施例中,该装置包括用于从分离通道收集未转向的流的储罐。
来自分析区的流输出和来自分离通道的未转向的流可以在储罐中被收集在一起。
在储罐中的组分可被收集用于进一步使用和分析。
本发明的装置允许流体流过分离通道、流分离器和检测区。流通过流体装置(如微流体装置)的建立是本领域技术人员众所周知的。例如,流体流可以通过注射泵提供,这些注射泵是用于各种流体通道的储罐。可替代地,流体流可通过流体到装置的重力自动加料来建立。在另一个替代方案中,流体流可通过例如使用注射泵从该装置的流体出口吸引液体穿过该装置来建立。
本发明的装置可以结合或使用这些不同流动系统中的一种或多种。
本发明的装置可使用标准光刻技术部分地制备,诸如在此所述的。
流体装置的通道表面可以适配为防止组分粘附到表面。因此,在一个实施例中,通道表面限制或防止组分被吸收到表面上。
在一个实施例中,流体装置内的通道是亲水性的或疏水性的。本发明的诸位发明人已经发现,使用亲水性通道表面,特别是在检测区,防止疏水组分(例如疏水蛋白)的吸收,从而改进该装置中的组分的分析。类似地,可以使用疏水性通道以防止亲水性组分的吸收。
特别地诸位发明人已经发现使用亲水性或疏水性通道表面在对组分进行标记和变性的阶段是有益的。在标记步骤中产生的不溶性物质的量被最小化。
亲水性通道可以使用本领域技术人员熟悉的技术制备。例如,当从pdms制备装置中的通道时,该材料可进行等离子体处理以使表面是亲水性的。在此,等离子体处理在通道的表面上产生亲水性的硅烷醇基团。此类技术是由tan等人(生物微流体(biomicrofluidics)4,032204(2010))描述。
在一个实施例中,在微流体装置中的通道,如在检测区的通道,具有亲水性或疏水性表面。
在一个实施例中,在微流体装置中的通道,如在检测区的通道,在其表面上具有羟基。在一个实施例中,在微流体装置中的通道,如在检测区的通道,在其表面上具有硅烷醇基。
流内标记
在本发明的另一个方面中,本发明的诸位发明人已经建立了一种用于标记在流体装置中、并且更具体地流体流内的组分的方法。这些组分以跨过层状第一和第二流体流的非均匀分布提供。如先前描述的,组分跨过第一和第二流体流的分离可以在其中该组分保持处于其原状态的条件下进行。一旦分布,这些组分随后可被标记用于后续分析。
该标记方法包括以下步骤:
(iii)提供該组分跨过第一和第二接触流(如第一和第二层流)的分布;
(iv)使该第一流体流的至少一部分、该第二流体流的至少一部分、或该第一流体流和该第二流体流的至少一部分转向,其中该转向的部分包括该组分;
(v)标记该流体流的转向的部分中的组分;并且任选地
(vi)分析该流体流的转向的部分中的该组分。
在一个实施例中,该方法包括以下准备步骤:
(i)在一种第一流体流中提供该组分;
(ii)使该流体流与一种第二流体流接触,如此以致产生层流;
以及步骤(iii)是允许该组分加入该第二流体流,从而获得该组分跨过该第一和第二流体流的分布。
在一个实施例中,步骤(iv)是使该第二流体流的至少一部分转向。
在一个实施例中,步骤(vi)存在。
本发明的诸位发明人已经发现,当该标记步骤向感兴趣的组分引入荧光标记物时该标记和分析步骤是最有效的。当标记物是共价标记物时标记也是最有效的,因为这消除了改变的组分浓度和构象模体对染料的结合亲和力的影响。还有利的是标记所有适当的反应性基团,不管组分序列、结构或浓度。当标记是快速的,并且在微流体实验的秒到分钟的时间尺度上达到完成(例如,在定量水平上)时,它也是最有效的。
本案描述了使用邻苯二醛(opa)用于产生标记的组分。
本发明的这方面的其他实施例如以上对于分析组分的方法描述的。
本发明的示例性方法和装置
本发明提供了用于分离和分析流体流中的组分,优选使用在此描述的微流体装置的方法。参照附图,以下陈述的是本发明的各种实施例的描述。
本发明的装置是用于分离并检测在混合物中的组分。该装置包括用于第一和第二层流的分离通道,并且该分离通道与下游流分离器流体连通。提供了一个检测区,该检测区是在下游并与该流分离器流体连通。该分离通道被适配为允许接触流(如层流)之间的组分的横向运动,并且该流分离器被适配为使该第一流体流的一部分、该第二流体流的一部分、或该第一流体流和该第二流体流的一部分从该分离通道转向。该检测区被适配为允许分析在该检测区的流体通道中的组分。
图1(a)插图示出的是根据本发明的一个实施例的微流体装置的示意图。该装置在图13中进一步详细示出。该装置适合于通过扩散方法分离组分。该装置包括一个分离通道1,该分离通道与一个下游流分离器7流体连通,该下游流分离器与一个下游检测区9流体连通。
该装置设置有一个分离通道1,该分离通道由一个上游第一流体流通道2和一个上游第二流体流通道3供应。该第一和第二流动通道在接合点4处结合。该第一和第二通道分别是由上游储罐5和6供应。该第一储罐5提供含有组分的流体,任选地与其他组分一起,例如作为多组分混合物的一部分。允许第一流体离开储罐并沿着第一流体通道流动。在接合点4处,允许第一流体流接触第二流体流,该第二流体流是从第二储罐6经由第二流体通道提供的。
第一和第二流体流可能在分离通道1中发展成层流。随着流向下传递到分离通道1,允许在第一流体流中的组分扩散到第二流体流中。组分或不同尺寸(不同的流体动力学半径)在不同速率下扩散,从而产生跨过第一和第二流体流的扩散分布。相比较大的组分,较小的组分将更快速地朝向在通道壁的第二流体流的边界扩散。
如在此所述的,第一和第二流体流通道的接合点4可以是具有大截面的通道,其随后发展成下游小截面通道(在图13中未示出,但是在图5(a)所示的装置中是可见的)。
在分离通道1的下游端,提供了一个流分离器7。该流分离器使该第一或第二流体流的一部分、或该第一和第二流体流二者的一部分转向。图13的流分离器旨在使该第二流体流的一部分,并且更具体地说,与较小尺寸的组分相关联的第二流体流的一部分(即已经更快速地朝向在通道壁的第二流体流的边界扩散的那些)转向。
流分离器7被跨过第二流体流的一部分放置以收集该第二流体流的一部分。转向的流进入下游检测区9的检测通道8中。
该转向步骤典型地在第一流体流中的组分已扩散到在通道壁的第二流体流的边界之前进行。因此,该组分的扩散分布跨过第一和第二流体流是非均匀的(因为该组分还没有达到跨过第一和第二流体流的平衡分布)。
层流的剩余部分被收集并允许经由收集通道11流至下游出口储罐10。
检测区9包括一个检测通道8,该检测通道与上游流分离器7流体连通。该检测通道8也与一个上游标记通道12流体连通,该上游标记通道在接合点13处结合该检测通道8。该标记通道从一个上游标记储罐14供应。标记混合物,任选地含有变性剂,在标记储罐14中提供并且允许经由标记通道12在接合点13处结合检测通道中的流。以此方式,标记试剂可被提供到从分离通道1转向的流(转向的流)中。
标记流结合进入检测通道8并且标记试剂标记该组分。允许标记流和转向的流混合足够的时间以允许组分的标记。在检测通道8中的流体然后在检测区9的分析区15中进行分析,例如使用荧光光谱法。一旦分析完成,允许在检测通道中的流体离开检测区9并收集在下游出口储罐10中。检测通道8结合收集通道11,从而重新组合来自分离通道1的流体流。
该装置的又另一个改变示于图12中,其中检测通道8与上游标记通道12和上游变性通道18流体连通。标记和变性通道12和18中的每一个分别在接合点13和19处结合检测通道8。变性通道的接合点19是在标记通道13与检测通道8的接合点的上游。检测通道8中的转向的流体首先与从变性通道18提供的变性剂接触,从而使在转向的流中的组分变性。该流然后与来自标记通道12的标记流接触,从而标记该流中的(变性的)组分。接合点12和18之间的距离足以允许流体流中的组分的完全变性。
在其他实施例中,在标记流中一起提供变性剂和标记物,如在图7和13中所示。
图1(a)和图13的装置的改变示于图7(a)中。在此,检测通道7具有在接合点13的下游的混合区。该混合区允许标记材料与来自分离区的转向的流混合足够的时间以允许在检测区9的分析通道15中分析之前标记。
图1(a)和图13的装置的另一个改变示于图10中。在该装置中,将来自分离通道1的流的一部分如前所述转向、变性并在单独的步骤中标记,并且如上所述分析。
该流的剩余部分被收集作为进一步分离的多个流。在这些附加转向的流中的每一个中的组分也被标记并进行分析,如关于转向的流所描述的。以这种方式,在分离通道1中的流体流被分离成三个流,其中这三个流中的每一个具有组分的不同混合物,这代表具有不同扩散特性的组分。流中的所有组分随后进行分析,以其分离的形式。因此,本发明的装置也用于分离和完全分析在第一流体流中提供的所有组分。
本发明的另一个装置在图14中示出。该装置适合于通过电泳方法分离组分。该装置包括一个分离通道1,该分离通道与一个下游流分离器7流体连通,该下游流分离器与一个下游检测区9流体连通。
该装置设置有一个分离通道1,具有大截面1a的区域和小截面1b的下游区域,其是由一个上游第一流体流通道2与上游第二流体流通道3a和3b供应的。该第一和第二通道在接合点4处结合。该第一和第二通道分别是由上游储罐5和6供应。该第一储罐5提供含有组分的流体,任选地与其他组分一起。允许第一流体离开储罐并沿着第一流体通道流动。在接合点4处,允许第一流体流接触第二流体流,该第二流体流是从第二储罐6经由第二流体通道3a和3b提供的。
在流体接合点处使用大截面通道与在接合点处的减少的滞留相关。参见,例如,pct/gb2013/052757。
第一和第二流体流可能在分离通道1中发展成层流。第二流体流被设置在第一流体流的任一侧。电极16和17被设置在分离通道1的任一侧。电极与电源(未示出)电连通。在使用中,电极提供跨过分离通道1的电场,如分离通道1的小截面区域1a。
随着流向下传递到分离通道1,在第一流体流中的组分响应于所施加的电场被偏转到第二流体流中。偏转的方向和程度取决于在第一流体流中的一种或多种组分的电荷和电荷与尺寸之比。
不同电荷的组分在定向方向上被偏转,朝向电极16亦或朝向电极17。其电荷与尺寸之比不同的(以及具有相同的电荷)的组分以不同的量被偏转到第二流体流中。
与具有较小电荷和/或较大尺寸的组分相比,具有较高电荷和/或较小尺寸的组分将在较大程度上朝向在通道壁的第二流体流的边界被偏转。
在分离通道1的下游端,提供了一个流分离器7。该流分离器使该第一或第二流体流的一部分、或该第一和第二流体流二者的一部分转向。图13的流分离器旨在使该第二流体流的一部分,并且更具体地说,与具有特定电荷(即朝向电极16吸引的那些)并具有较小电荷和/或较大尺寸(即朝向在通道壁的第二流体流的边界最少快速被偏转的那些)的组分相关联的第二流体流的一部分转向。
流分离器7被跨过第二流体流的一部分放置以收集该第二流体流的一部分。转向的流进入下游检测区9的检测通道8中。流分离器7和检测通道8在图5(b)中更详细示出。
流分离器7被跨过第二流体流的一部分放置以收集该第二流体流的一部分。转向的流进入下游检测区9的检测通道8中。该转向步骤典型地在第一流体流中的组分已被偏转到在通道壁的第二流体流的边界之前进行。
层流的剩余部分被收集并允许经由收集通道11a和11b流至下游出口储罐10。
检测区9包括一个检测通道8,该检测通道与上游流分离器7流体连通。该检测通道8也与一个上游标记通道12流体连通,该上游标记通道在接合点13处结合该检测通道8。该标记通道从一个上游标记储罐14供应。标记混合物,任选地含有变性剂,在标记储罐14中提供并且允许经由标记通道12在接合点13处结合检测通道中的流。以此方式,标记试剂可被提供到从分离通道1转向的流(转向的流)中。
标记流结合进入检测通道8并且标记试剂标记该组分。允许标记流和转向的流混合足够的时间以允许组分的标记。为此目的在分析区15的上游设置混合区。在检测通道8中的流体然后在检测区9的分析区15中进行分析,例如使用荧光光谱法。一旦分析完成,允许在检测通道中的流体离开检测区9并收集在下游出口储罐10中,从而重新组合来自分离通道1的流体流。
其他参数选择
在此明确披露了以上所述的实施例的各个和每个相容性组合,就像单独地且明确地叙述各个和每个组合一样。
鉴于本披露,本领域技术人员将清楚本发明的多个不同的其他方面和实施例。
在此使用的“和/或”应当理解为明确地披露了两个具体特征或组件中的每一个(具有或不具有另外一个)。例如,“a和/或b”应当理解为明确地披露了(i)a、(ii)b和(iii)a和b中的每一种情况,就像每一种情况在此单独地给出一样。
除非上下文中另外指出,否则上述列出的特征的描述和定义并不限于本发明的任何特定方面或实施方式,并且等同地应用于所描述的所有方面和实施例。
现在将仅借助于实例并参照以上所述的附图来说明本发明的某些方面和实施例。
实验
批量定量标记实验
牛血清白蛋白(产品#a7906)、溶菌酶(#62970)、β-lactogloublin(#l3908)、十二烷基硫酸钠(#71725)、碳酸氢钠(#8875)、hepes(#h3375)和邻苯二醛(#79760)是从西格玛奥德里奇(sigmaaldrich)获得。十水碳酸钠是从东安格利亚化学公司(eastangliachemicalcompany)(产品#1353)获得。β-巯基乙醇是从赛墨科技公司(thermoscientific)(产品#35602)获得。
研究了各种opa:bme比率,并且证明1:1.5是最佳的。还研究了各种缓冲条件。在ph为9.5和10.5之间,使用高离子强度(100至500mm)的碳酸盐缓冲液,获得了最佳定量检测灵敏度。标记反应耐受各种缓冲液。最初认为含有伯胺的缓冲液(如tris)将干扰定量标记反应。然而,最近的工作已显示tris-型缓冲液并不是问题。
研究了各种标记条件。最初的实验确定为了确定在nm到高μm范围内的蛋白质浓度,6mmopa、9mmbme的最终opa/bme浓度是最佳的。检验了各种变性剂和表面活性剂,包括20%-40%dmso、30%-40%etoh、2%sds,单独地或与0.5%-5%吐温-20组合。
12mmopa、18mmbme、4%sds、以及200mm碳酸盐,ph9.5-10.5的标准标记溶液被用于电泳和扩散分离和检测实验。典型地提前制备该溶液并且以与感兴趣的蛋白质溶液的1:1体积比混合。,如果避光的话,标记溶液可以储存长达1周而没有性能的可检测的损失。浓度改变的bsa、溶菌酶和β-乳球蛋白溶液在5mmhepes缓冲液,ph7中制备并与以上标记溶液混合。
对于批量检测和标记实验,每个条件的三次重复装入低蛋白结合、半区96孔板(康宁,产品#3881),并用粘合铝密封剂片材(柯仕达(costar)产品#6570)覆盖。端点荧光测量在bmglabtechfluostaroptima读板机上,采用350+/-10激励和440+/-10发射滤光器进行。
微流体装置制造和操作
微流体扩散装置是使用标准光刻方法(参见kim,p.等人生物芯片杂志(biochipjournal)2,1-11(2008))制造的。简略地,在autocad中设计装置,二元掩膜印刷在乙酸酯片上(microlitho),具有对应于在微流体装置中的预期通道的透明区域,和对应于背景的黑色区域。掩膜-待流延的装置的正影像-是通过以下制备的:将25μm的microchemsu-83025永久环氧负性光致抗蚀剂旋涂到硅晶片上,在光致抗蚀剂的顶部铺设该掩模,用校准的uv光交联暴露的环氧树脂,并用丙二醇单甲醚乙酸酯(pgmea)显影剂(microchem)最终去除未交联的聚合物的区域。
装置在聚二甲基硅氧烷中流延。pdms弹性体和固化剂(道康宁(dowcorning),产品#184)以1:1w/w混合。重要的是确保混合完全:2-5分钟的人工搅拌对于适当固化的弹性体的性能是重要的。当黑色装置是希望的时,将约20mg的碳纳米粉末(西格码(sigma),产品#633100)加入并充分混合。大团块的纳米粉末通过在eppendorf5804r离心机中以3,000rpm离心10分钟沉降。将混合的弹性体和固化剂倒入掩膜上,通过真空干燥约10分钟除去气泡,并且将装置在70℃下烘烤60-75分钟。一旦冷却,将装置关掉,并且用0.75mmharrisuni-core打孔器在入口和出口处打孔。在用胶带除去碎屑后,将它们使用electronicdienerfemto等离子体结合机(electronicdienerfemtoplasmabonder)等离子体结合到赛墨科技公司(thermoscientific)76×26mm载玻片(目录#8037)上。将结合的装置在70℃下烘烤10分钟。
对于基于电泳的分离实验,电极根据公开的程序在单个步骤中制造(参见herling等人)。简略地,电极通道用25μmpdms立柱描绘。在装置结合之后,将它们在热板上加热到78℃,并将低熔点焊料(由51%in、32.5%bi、16.5%sn组成的inbisn合金,conroelectronics)推入电极入口。熔融焊料的高表面张力将它局限于电极通道,同时确保在电极和缓冲水溶液之间的接触。
在装置中的流是使用nemesys注射泵控制的。明场和荧光图像是使用zeissaxioobserver显微镜,配备有365nmcaimoptoled(photometrics),和用于荧光图像的chromo49000dapi过滤器获得的。使用了2.5x、5x、10x和20x物镜。使用了在100ms和1000ms之间的曝光时间。荧光图像在黑暗中拍摄,并且通常是平均10个单独的图像的结果。在某些情况下,图像被合并以增加信噪比。图像是通过减去以相同的曝光设置拍摄的平场图像校正的背景。
微流体实验
牛胰岛素是从seralab(产品#gem-700-112-p)购买。胰岛素含有0.6%w/wzn2+并且按收到时原样使用。剩余的实验方案在下面详细描述。
电泳psl实验:将10mg/mlbsa和10mg/ml溶菌酶各自溶解在5mmhepes,ph7中。向溶液中加入100μm罗丹明6g(西格马)。然后将溶液通过0.22μm注射器过滤器过滤。制备含有11mmopa、16.4mmbme和3.63%sds在180mm在ph9.5下的碳酸盐中的标记溶液。将溶液装入装置入口的每一个中,并且然后将电极焊接到电线上,这些电线进而连接到一个dc电压源。罗丹明6g示踪物(不干扰opa荧光)的位置被用来验证装置对准。检测通过检查在-5v、0v和5v下罗丹明光束的变化的位置验证。经验证的成功检测,当电压在-10和8v之间变化时,在观察区测量了opa荧光强度。荧光在混合界面处的形成也被可视化。重要的是避免施加高电压持续仅短的时间段,以避免由于电解在电极处的气泡形成。对于单独的装置,装载溶菌酶溶液来重复相同的程序。
标记
在此所述的组分的分离和检测使用微流体的快速空间隔离可能性。使用这种方法,未修饰的组分,如生物分子,根据它们的固有特性像尺寸或电荷在空间上分离。这些组分然后可以被收集并随后检测。该检测步骤可以包括将分离的组分暴露于一组新的条件,这些条件促进与标记物的快速、完全的反应和后续定量检测。
对于标记方法重要的是两个、先前无关的想法的融合。第一个是组分与潜在共价标记物(lcl)的反应,该潜在共价标记物是仅在与该组分上的相关官能团反应时迅速变成荧光的分子。第二个是快速变性步骤,其使所有相关官能团暴露于溶剂,从而加速并且确保与该潜在标记物的完全反应。
潜在共价标记方法在此是关键的,因为它消除了对于在标记反应后的样品纯化的要求。因为只有标记的底物是荧光的,未反应的标记前体并不需要被去除。这允许标记和检测过程直接紧接着在微流体芯片上的分离方案。此外,该标记反应的共价性质确保所有物种被永久修饰,使得相对检测灵敏度不依赖于例如染料结合亲和力的差异。
可以设想两类潜在共价键标记物。在第一个中,与底物部分的反应除去淬灭其荧光的最初存在于标记物上的基团。在第二个中,荧光团本身在标记反应过程中形成,例如通过延伸的共轭体系的形成。
微流体方法的限制之一是微流体装置的流体分析所要求的快速反应动力学。对上面讨论的第一种方法进行了探讨,期望荧光团通过共轭体系的延伸的形成可以充当快速、装置内反应的足够的驱动力。
对各种这样的荧光化合物进行了研究,并且采用邻苯二醛(opa)取得了期望的结果。
用opa标记
在1971年,roth等人发现,如果opa在硫醇如β-巯基乙醇(bme)的存在下与分离的氨基酸反应,则形成蓝色荧光产物(罗斯分析化学(analyticalchemistry)43,880{882(1971))。观察到的快速动力学然后指引作者开发了在离子交换柱中在分离后用于在线氨基酸修饰的相关技术(roth等人,色谱杂志(journalofchromatography)83,353-356,(1973))。这种修饰技术随后被证明在的检测灵敏度和室温反应快速性方面优于更多标准的肽修饰系统(如使用茚三酮的那些)(benson等人,美国科学院院报(pnas)72,619-622(1975))。描述了随后的努力,在毛细管电泳之前、期间或之后使用opa作为氨基酸或生物分子衍生化试剂(oguriet等人,色谱杂志(journalofchromatography)a787,253-260(1997);jacobson等人分析化学(anal.chem.)1994,66,3472)。根据诸位发明人的知识,没有使用opa作为在微流体空间分离之后的装置内衍生化试剂的报道的实例。
方案1-在碱性条件下的标记机制,改编自sternson和garcia-alvarez-coque等人分析生物化学(anal.biochem.)144,233-246(1985);garciaalvarez-coque等人分析生物化学(anal.biochem.)178,1-7(1989))。
在碱性条件下的opa反应机制如方案1中所示。opa二醛与半缩醛和硫代半缩醛形式平衡。二醛与蛋白质表面上暴露的伯胺反应,并失去水,从而形成高度反应性的亚胺。bme硫醇盐对亚胺的攻击释放用于攻击侧链醛的仲胺,闭合5元环。该反应在共轭的延伸和水的失去的情况下是不可逆的。
检测灵敏度
使用opa用于蛋白质浓度的纳克检测灵敏度已经在文献中报道(zawieja等人分析生物化学(analyticalbiochemistry)142,182-188(1984))。文献研究使用在<nl样品体积中μm的蛋白质浓度,但是,并允许至少30分钟的反应时间获得这种水平的灵敏度。
使用opa用于线性蛋白浓度测定的检测灵敏度是在微流体衍生化的约束下确定的。
微流体装置的设计如图1中(a)的插图所示,并相对于示例性方法和装置如上所述。该反应在底流进行,并且因而是处于稳态,但总反应体积(定义为在任何时间点标记蛋白的总体积)是0.38μl。荧光强度在检测区测定,如图1(a)中的细长长方形所示。在该实验中,荧光强度在染料和蛋白质流束在y接合点处最初接触之后18秒测定。
如图1(a)中所示,当bsa在微流体装置中用opa标记时,opa荧光强度随bsa浓度线性下降到15nm。该检测限可以通过在标记反应之前或过程中使用合适的、低背景条件使蛋白质变性,如将在下面讨论的,或者通过增加装置路径长度来进一步减小。
用于这些实验的装置具有25μm的路径长度,并且最高达100μm的路径长度是可达到的而不改变制造工艺。预期对于100μm路径长度的装置的灵敏度被减小到3.75nm。相比之下,吸收检测灵敏度示于图1(b)中。a280和bsa浓度之间的线性关系仅对于1mm路径长度对于大于585nm的bsa浓度获得。转换成在微流体装置中使用的25um路径长度,预期23.4μm的检测限。因此,装置内衍生化方法在确定未修饰蛋白的浓度时是比吸收法更敏感超过1,500倍。
即使在当前配置中,观察到蛋白质浓度和荧光强度之间的线性关系下降到0.38ng的蛋白质,这是与文献值可比较的结果(zawieja等人分析生物化学(analyticalbiochemistry)142,182-188(1984)),但对于约1,000倍更低的绝对浓度的蛋白质,以及约100倍更少的反应时间。
定量标记
在此描述的标记方法的另一个重要组成部分是认识到,通过使感兴趣的组分(例如蛋白质)变性并且暴露所有相关的官能团(例如氨基)用于反应,可达到完全标记。使用基于opa的标记,荧光强度可以定量地用于确定蛋白质浓度。变性可以发生在标记之前或期间。
已经探索了各种条件,并且对于若干变性和标识混合物,蛋白质浓度可以定量地与opa荧光强度相关,如以下所讨论的。图3示出了在以下描述的扩散和电泳分离和检测方法中使用的条件。
如图2中示出,将不同浓度的bsa、溶菌酶、和β-乳球蛋白,具有广泛不同的等电点(iep)、分子量、和一级序列的三种模型蛋白质,与标记混合物混合为6mmopa、9mmbme、2%十二烷基硫酸钠(sds)、和100mm碳酸盐,ph10.1的最终标记浓度。蛋白质一级序列被用于将最终蛋白质浓度转换为伯胺浓度,并且绘制对于三种蛋白质的opa荧光强度和伯胺浓度之间的关系。线性回归产生了0.99的相关系数。sds在标记溶液中的存在略微增加了来自未反应的染料的背景。在本实验中使用的高离子强度下,sds胶束形成复杂多样的结构(almgren等人,胶体与界面科学杂志(journalofcolloidandinterfacescience),202,222-231(1998)),预期其中的一些将散射未反应的染料背景荧光。
定量标记方法可以用于确定未知的肽浓度。类淀粉蛋白-β(1-42)(aβ(1-42))没有色氨酸残基和1,4001/mcm的低消光系数(walsh等人,febs杂志(febsjournal),276,1266-1281(2009))。显示出产生对于aβ(1-42)的聚集的可重现的动力学数据的仅有的文字记载的程序涉及重组肽的表达、纯化、和动力学分析之前立即进行纯化的肽的sec过滤以除去之前形成的聚集体(walsh等人,febs杂志(febsjournal),276,1266-1281(2009));hellstrand等人,acs化学神经系统学(acschemicalneuroscience),1,13-18(2010))。然而,这常常导致难以通过吸收检测的低浓度的纯化的单体。因此,获得对于aβ(1-42)的可重现的动力学数据额外地取决于对比仅从单个批次的纯化的单体制备的样品,使得每一个样品将具有相同的浓度相对误差。
图3示出了在此提出的定量标记方法可以如何用于确定纯化的aβ(1-42)肽的浓度。在图3(a)中,伯胺浓度和opa荧光强度之间的线性关系是通过测量已知浓度的β-乳球蛋白的荧光强度测定的。重要的是,因为该标记反应是定量的,对于一组给定的测量条件这种关系只需要测定一次。
使用相同的实验条件对未知浓度的aβ(1-42)测定荧光强度。在这两种情况下误差条是三次重复的标准偏差。因为蛋白质伯胺浓度和opa荧光强度之间的线性关系,a(1-42)伯胺浓度从而是从观察到的荧光强度确定的。图b显示了在对稀释因数校正之后,对于每个系列稀释确定的相应蛋白质浓度。在整个浓度系列获得约27μm的稳定值。可以使用这种方法以便在进一步的生物物理研究之前快速地且准确地确定aβ(1-42)和aβ(1-40)浓度。此外,可以使用类似的方法以确定流体流中的任何蛋白质的浓度,条件是一级氨基酸序列是已知的。
证明了在微流体实验中在快速的时间尺度下定量标记组分的能力。在文献中存在极少opa反应动力学的报告(yoshimura等人,分析生物化学(anal.biochem),164,132-137(1987);wong等人,美国化学会志(j.am.chem.soc.),107,6421-6422(1985))并且没有全长蛋白质的改性的速率,或在变性剂存在下的opa反应的速率的报告。使用图7(a)中示出的装置,发现使用以上识别的含有sds的变性条件,bsa改性在约1s内进行至完成;在蛋白质和定量潜在共价标记溶液混入并退出等待环路后,荧光强度是恒定的。代表性的图像在图4中示出,其中荧光强度的恒定性用强度曲线示出。
以上描述的是一种新颖的定量潜在共价标记方法,该方法允许在流体装置中在横向分离和转向之后用荧光染料定量地标记组分。因为该标记是定量的,蛋白质浓度可以直接从荧光强度来确定。这种方法将测量和检测过程分开使得标记物的存在不影响测量和分析。下面讨论的是将这种标记应用到使用电泳和扩散分离的分离和检测的方法中。
扩散分离
当流体被限制在毫米长度尺度,流是层流的,而不是对流的。因此,当流体的两个相邻的流束相遇时,如在y-或t-接合点,流体的层之间的唯一混合是由于扩散(whitesides自然(nature)442,368-373(2006);squires等人现代物理评论(reviewsofmodernphysics)77,977-1026(2005))。感兴趣的物种的扩散系数和流体动力学半径因此是从它的空间分布可取得的。在此描述的是一种用于扩散分离的物种的衍生化和定量的装置。代表性的扩散装置在图1(a)中示出。该装置具有两个流入口。参看图1,缓冲液被装入一个入口(左下),并且蛋白质被装入另一个入口(左上)。仅扩散到缓冲液流中的蛋白质可被转向和标记。沿扩散通道的长度行进后,从缓冲液和蛋白质流之间的层流边界已经扩散至少33μm进入缓冲液流中的蛋白质被分离(转向)并且随后在流体装置内与标记混合物混合。标记混合物包含一种标记物(除非另外指出,opa)和一种变性剂(除非另外指出,sds)。
标记蛋白质的荧光强度在矩形观察区域中进行测量。未标记的蛋白质也溶解,所以它不会形成不溶性聚集体,例如,当它与碱性标记溶液相结合时,似乎它越过蛋白质等电点。
准确的流体动力学半径的提取-尤其是对组分的不均匀混合物-要求实验数据与对于已知尺寸的组分所产生的模拟空间扩散分布的比较。在此描述了这种比较,并且参考pct/gb2013/052757。
胰岛素
作为扩散方法的初始示范,研究了单体和小低聚物在胰岛素聚集反应中的消耗。还研究了在胰岛素六聚化中ph诱发的变化的影响。胰岛素是观察到的形成体外类淀粉蛋白原纤维的第一系统之一(waugh美国化学会志(j.am.chem.soc.)68,247-250(1946))。胰岛素已经充当一种用于低聚和类淀粉蛋白聚集的便利生物物理模型系统。胰岛素随ph变化形成明确定义的二聚体、四聚体和六聚体(nettleton,e.j.等人生物物理学杂志(biophysicaljournal)79,1053至1065(2000);whittingham等人分子生物学杂志(journalofmolecularbiology)318,479-490(2002))。
其类淀粉蛋白原纤维的原丝结构已经通过低温电子显微镜测定(jimenez等人胰岛素原纤维组装分子基础(molecularbasisforinsulinfibrilassembly)美国科学院院报(pnas)99,9196-9201(2002)),以及对通过x射线晶体学的交叉骨刺(cross-spine)关键的中心段的结构(ivanova等人美国科学院院报(pnas)(2009))。最近的工作(knowles等人,科学(science)326,1533-1537(2009);cohen等人分子生物学杂志(journalofmolecularbiology)421,160-171(2012))允许从间接监测的tht荧光数据提取反映聚集过程中的离散步骤的微观速率常数(levine等人蛋白质科学(proteinscience)2,404-410(1993);biancalana等人生物化学与生物物理学学报(biochimicaetbiophysicaacta)1804,1405-1412(2010))成熟类淀粉蛋白原纤维的形成。用于直接追踪在类淀粉蛋白聚集反应中的单体和小低聚物的变化群体的现有的实验方法扰乱在观察下的瞬态过程,通过转移进入气相(nettleton,e.j.等人,生物物理学杂志(biophysicaljournal)79,1053-1065(2000)),外源性标记和稀释(73),或长测量时间(schuck分析生物化学(anal.biochem.)320,104-124(2003);mok等人方法(methods)54,67-75(2011))。
在此描述的扩散分离和检测方法是能够不间断地追踪单体和小低聚物消耗的第一直接实验方法。
胰岛素聚集通过在60℃下在静止条件下培育2mg/ml牛胰岛素启动。如在图7(b)中所示,原纤维化的过程通过tht荧光的增加实时监测。将来自没有添加的tht的样品的等分部分在对应于在t=0未加热的单体,第一可检测的tht荧光的增加,滞后时间,以及平衡相的时间点除去。
将等分部分装入在图7(a)中示出的扩散装置的蛋白质入口。
将来自分离通道的最小物种转向到检测区。转向后,将转向的流首先与潜在标记混合物(12mmopa,18mmbme,4%sds,200mm碳酸盐ph10.3)混合。然后通过荧光光谱法检测标记的胰岛素物种。
图7(b)用对应的原纤维化曲线覆盖了对于这些时间点的归一化的荧光强度值。这被认为是对在类淀粉蛋白聚集反应中的单体和小低聚物的消耗的首次非干扰直接实验分析。
本发明的分离和分析方法也被用来研究ph-诱发的胰岛素低聚。在低ph下,胰岛素主要作为二聚体(61)存在。在中性和碱性ph下,在zn2+的存在下,胰岛素作为六聚体(76)存在。因此,在ph2.0和10.5下制备2mg/ml牛胰岛素溶液。将这些溶液装入在图1(a)中示出的装置的上部蛋白质入口。然后使扩散了33.33μm或更多的物种转向,并随后标记并测量该流的荧光强度。胰岛素用以上使用的opa标记混合物标记。分离、转向、标记和检测步骤所有都在稳态流动条件下在单个装置上进行。
在ph2和ph10.5所观察到的荧光强度如图7(c)中绘制。有吸引力的是推测在碱性ph下的较低的相对荧光强度对应于胰岛素六聚体。
绝对流体动力学半径
在图7(b)和(c)中记录的数据使用相对荧光强度的差异来指示不同比例的小物种,这些小物种扩散足够远以被标记。
虽然这些数据的确产生关于样品组之间的相对差异的定性信息,额外地有可能使用基于扩散的分离和检测方法以获得绝对流体动力学半径,通过将已知的数值模拟算法适配为扩散光谱法的一部分(见pct/gb2013/052757)。
对于已知流体动力学半径的球形颗粒,模拟生成了预期的颗粒密度分布—称为“基函数”。此模拟以对应于理论预测的和实验观察的“帽子”函数的初始分布将已知尺寸的颗粒跨越“喷嘴”随机分布。允许颗粒通过扩散通道传送,采取布朗运动随机步骤,这些步骤在整个通道中导致非匀速分布(lauga等人,urlhttp://arxiv.org/abs/cond-mat/0501557),并最终到达检测区。
然后通过将对于每个颗粒到达观察区之前的每个时间点获得的分布求和来模拟该尺寸的颗粒的稳态分布。在扩散光谱法中然后使用这些基函数通过最小二乘拟合算法来评估已知尺寸的颗粒对实验观察到的空间扩散分布的相对贡献。在将此已知的方法应用于在此描述的方法中存在两个相关的问题有待考虑。空间扩散分布典型地不在分离区中测量。对此的原因是组分有可能未被标记,并且因此是不容易检测的。在本发明的一些实施例中这些组分可以是可检测的并且因此可以在分离步骤期间,例如在分离通道中,测量扩散分布。
在扩散分布不能在分离步骤期间测量时,该组分在转向后稍后进行分析。例如,转向的组分可以在转向后被荧光标记并且实验可观察的是在检测区内的整体荧光强度。因为该标记步骤是定量的,有可能将整体荧光强度与观察区内的组分浓度相关联。然后,一旦对于使用的设置构建了校准曲线,流体动力学半径可以通过将表观组分浓度与已知装载的组分浓度相关联(条件是已经装载了仅一种单一组分)理论上获得。然后可以将实验观察的比率与来源于模拟的类似颗粒密度比率相进行比较,在该模拟中允许已知流体动力学半径的颗粒在整个分离通道中扩散。
然而,依靠绝对荧光强度在实验上是有问题的,因为绝对荧光强度和因此获得的表观浓度将取决于图像设置而变化,并且实际上甚至取决于光学对准的精密差异。这种变化性可以通过包括附加的实验和理论数据点:如在图6中示出的当组分被装载在两个入口时观察到的荧光强度(或颗粒密度),而不是对这些因素的内部校正来消除。这些值的比率给出了已经扩散足够远以被标记的总装载的组分浓度的部分,其可与对应的模拟结果对比以得到样品流体动力学半径的精确值。
这些模拟的结果在图8和9中示出。在图8中,模拟了具有已知流体动力学半径的颗粒的扩散。流体动力学半径在0.5和50.0nm之间对数地隔开,与范围在小分子和大蛋白质复合物或聚集体之间的尺寸的物种的检测有关的尺寸方案。
模拟了组分在扩散通道的端部的稳态分布(之后感兴趣的物种被转向并任选标记和后续分析)。该模拟是基于一种系统,其中颗粒被装入一个装置入口和其中颗粒被装入两个装置入口的情况,如在图6中示出。这些模拟的结果是根据流体动力学半径颜色编码的并在图9中示出。如所预期的,颗粒的稳态分布不随流体动力学半径改变其中在两个流体入口均提供组分(在此,蛋白质):初始条件扩散的均匀性不产生在稳定状态下的颗粒分布的净改变。相反,对于其中仅在一个提供组分的情况下(图6中的顶部入口),在检测区的稳态分布预期地随流体动力学半径而变化。
基于在图6中示出的装置设计,物种必须扩散至少33μm以便通过下游流分离器转向。转向后该组分与等待环路中的标记溶液混合,并且该标记的组分然后通过荧光方法检测。因此,已经扩散足够远以被标记的物种的部分是其中在分离通道的端部并且处于稳态的那些是位于沿着通道的宽度在133和200μm之间,以0μm为蛋白质入口的远壁。
为了捕捉这个部分,对于其中在两个入口提供组分的条件,对颗粒分布曲线的相应区域进行整合。对于已知的流体动力学半径的每一个这些强度的比率给出了已经扩散足够远以被转向的总装载的组分浓度的部分。通过将实验观察的比率与模拟的那些对比,可以获得绝对样品流体动力学半径。
在图9中,展示了流体动力学半径对实验观察到的扩散比率的依赖性。主图是以双对数格式示出的,而插图是以线性格式示出的。最大的四个流体动力学半径给出了0的扩散比率(对于数值近似),所以将这些点从对数曲线图中移除。对于一阶近似,数据可以相当好地拟合到幂律:
f(x)=axb
其中a=1.13且b=0.22,具有0.97的r2,并且模拟结果的可预测性展示了实验观察到的扩散比率可以如何明确地与流体动力学半径相关。
然而,除非分析的样品是单分散的,预期以上技术产生平均流体动力学半径。来自复合蛋白质混合物的多种组分的流体动力学半径可以通过扩大实验条件的系列其中对物种扩散进行建模来获得。如在图10(a)中示出的,已经设计了选择并定量地标记扩散样品的多个部分的流体装置的变型。在此,来自分离通道的流被分离(转向)成三个相等的组分。
区域1对应于从组分流扩散最远(至少33μm)的组分。区域2收集组分的中间三分之一,以及区域3最后三分之一,或在远组分壁的66μm内的扩散室的端部处于稳态的组分。在此配置中,每个转向的流是分别变性的,并且然后流中的变性组分用含opa的混合物标记。
图10(b)示出了参考样品流体动力学半径对扩散比率1、2和3的依赖性。扩散比率如上计算。通过扩展理论处理以覆盖各种流速(在此显示的基函数对应于在扩散通道中37.5μl/hr的单个抽取速率),对于每个流体力学半径,有可能绘制扩散比率1、2、和3对流速的依赖性。这些基曲线对观察到的实验数据的相对贡献将用最小二乘拟合程序确定。这产生了感兴趣的不均匀样品中的流体动力学半径的清单。因为标记反应是定量的,可以确定实验样品中的每个流体动力学半径的相对丰度。
可溶性
在此提出的准备胰岛素聚集和六聚数据是定性的,并且它没有涉及强度的变化直接改变样品的流体动力学半径。对此的原因与引起流-速从选定值的不规则偏差的溶解度问题有关。如果含有组分的流被改变,例如在组分的标记过程中,使得越过等电点,然后该组分在组分和标记混合物相遇的层流界面处“冲出”。对应于图7的图c中示出的ph诱发的胰岛素六聚化实验的代表性图像在图11中示出。聚集蛋白的栓塞是最终可溶的,但在两个流束之间的混合区中存在沉淀的蛋白质阻碍了流并且导致不可预测的流速变化,这给出扩散曲线对流速的灵敏度,将使得从相对荧光强度变化提取定量流体动力学半径是有问题性的。
这个问题不是特别针对胰岛素并且当感兴趣的蛋白质在标记时越过它的等电点时对于多种系统已经观察到了同样的问题。
为了提出对于将与任何感兴趣的蛋白质系统工作的定量分离和检测方法的真正通用方法,溶解性问题已经以各种方式得以解决。
当胰岛素标记反应批量重复时没有形成不溶性物种。例如,在ph2胰岛素与等体积12mmopa、18mmbme、以及在200mm碳酸氢盐中4%sds,ph10.5迅速混合,并且所得溶液的ph为10.5。
因此,该溶液被有效地缓冲至以下ph,在该ph下单体胰岛素在分离中是可溶的,并且完全混合确实得到可溶性混合物。这表明当胰岛素用变性剂有效地溶解时,胰岛素在该ph附近受到保护免受聚集。假设这种类型的溶液在层流界面处冲出因为在蛋白质能够与清洁剂胶束结合之前在界面处的蛋白质溶液的ph能够变化,简单地因为h+浓度和胶束扩散系数的差异。
注意,当胰岛素溶液在室温下放置时,在天的时间尺度上观察到沉淀,尽管这个时间尺度与流体流标记实验无关。
因此,开发了包括分离变性和标识步骤的一种方法。在该方法中使用的装置是如图12(a)中所示地示出。在转向后,待标记的组分,如蛋白质,首先与变性剂完全混合然后随后与标记混合物混合。此外,在未转向的流中存在的未标记组分与变性剂混合以防止当它随后与装置出口附近的转向的流重新组合时该组分沉淀。
图12(b)示出了当在ph2的9.1mg/ml胰岛素与两种单独的变性和标记溶液在芯片上混合的结果。界面1对应于未标记蛋白质和变性剂的混合,界面2对应于变性蛋白质和标记混合物,并且界面3对应于未标记的蛋白质(其已经预先与变性剂混合)和标记且变性的蛋白质混合物的混合。当sds用作标记步骤之前的变性剂时,观察到在层流界面处的粗大的聚集体形成,其比当在单个步骤中进行的sds变性和标记时形成的暂时堵塞显著得多。虽然传统上认为sds溶解所有蛋白质,它还可以与带正电荷的残基,如赖氨酸和精氨酸,静电相互作用,形成疏水离子对。这种效果对于低于它们的等电点的蛋白质是特别相关的,并且减少胰岛素水溶性的胰岛素-sds相互作用已经明确地被报道(powers等人,生物聚合物(biopolymers),33,92-932(1993))。
考虑了多种其他离子和非离子表面活性剂(如tween-20和triton-x)连同不同的蛋白质变性机制(otzen,生物化学生物物理学报(biochim.biophys.acta.),1814,562-591(2011);otzen,生物物理杂志(biophys..j.),83,2219-2230,(2002)),碱金属和碱土金属盐(ahmad,生物化学杂志细胞生物学杂志(canj.biochem.cellbiol.),63,1058-1063(1985)),以及已经报道为使蛋白质变性的有机溶剂(brandts等人,美国化学会志(j.am.chem.soc.),89,4826-4838(1967);hirota等人,蛋白质科学(proteinscience),6,416-421(1997)),以及它们的组合(flockhart等人,胶体科学杂志(journalofcolloidscience),12,557-565(1957)),出于它们对在分析步骤过程中均溶解蛋白质的能力。表面活性剂和变性剂空间的研究已经揭示了存在多种条件,这些条件确保定量标记包括,例如,有机溶剂如etoh和sds的混合物产生类似于图2、4中示出的数据。已经显示了唯一一个条件用于在流中越过等电点可靠地溶解蛋白质,然而:在标记之前等体积分数的蛋白质和100%etoh。
用此条件观察到的改进的溶解性在图12(b)中示出。如在图12(c)中所示,这种情况不会导致定量标记。没有观察到伯胺浓度和荧光强度之间的强线性关系,并且溶菌酶溶液在批量标记实验中变得浑浊,这表明除了不完全标记外,检查的etoh混合物赋予的在低等电点的蛋白质的溶解性不是一般的。
最后,结合了分离变性步骤的扩散装置的空间实际制造要求使用若干伸长的非常长的、狭窄的通道,这些通道充当其中发生扩散混合的等待环路。这些通道的尺寸(20μm宽、25μm高、以及30mm长的数量级)可能难以制造并且在使用中形成微泡,例如当由于在填充过程中不利的润湿特性(monahan等人,分析化学(analyticalchemistry),73,3193-3197(2001))。微泡的存在严重影响达到的流速。由于在通道内的低于大气压的压力,微泡以溶液流过装置为代价生长,引起可变的低速率(kang等人,芯片实验室(labonachip),8,176-178(2008))。
在使用在图12(a)中示出的分离变性装置对于模型蛋白质(如上所述)获得绝对流体动力学半径的初始努力产生非物理扩散比率之后,研究了在一系列流速下对于已知尺寸的固有荧光小分子(荧光素)的扩散分布。图12(d)示出了,如预期的,在接合点处的“帽子函数”初始分布是如预期的,但该扩散分布没有随流速预期地变化。因此可以得出结论扩散通道内的不可预测的流率-很可能由在此复杂性的装置中微泡的存在引起-得到与预测扩散比率的偏差。
总之,图12证明不存在一组(广泛研究的)化学条件同时满足组分(尤其是蛋白质)的化学要求,溶解和变性,和可预测的操作的物理要求,而不牺牲其他因素如检测灵敏度。时间过程研究(在此未示出)证明在层流界面处冲出的蛋白质最终重新溶解。因此,在不存在与永久减小的水溶解性有关的条件像在图12(b)中示出的分离变性条件,问题不是真正缺乏溶解性而是当暂时聚集的蛋白质在混合接合点阻塞流时观察到的可变的流速。
在探索了多种混合接合点几何结构(数据未示出)并且发现堵塞行为没有变化之后,据信该效果是由于微流体流体行为的一个更基本的特性。无滑动边界条件得到在通道边缘的零速率和在整个通道的变化的速率分布(lauga等人,urlhttp://arxiv.org/abs/cond-mat/0501557)。据信在层流界面处的不溶性蛋白质,在发生扩散混合之前,沉积在低速的区域,堵塞装置。
为了检验这一假设并且理想地减轻上述的溶解性问题,本发明的诸位发明人目前正在开发三维分离和检测装置,其中组成部分将由染料和变性剂的鞘被垂直侧面包围,使得在层流界面处不存在低速区域。此外正在研究具有活性混合组成部分的微流体装置,如旋转磁珠或颗粒(stone等人,流体力学年鉴(annualreviewoffluidmechanics),36,381-411(2004);rida等人,分析化学(analyticalchemistry)76,6239-6246(2004);lee等人,芯片实验室(labonachip),9,479-482(2009)),以便在初始接触后迅速溶解蛋白质,并破坏可能发生的任何沉淀。
本发明的装置和方法允许基于组分的电泳和扩散特性分离这些组分,和这些组分的后续检测,任选地连同标记步骤。
在扩散用作分离方法时,有可能通过将实验“扩散比率”与对于已知流体动力学半径的物种模拟的相似比率相关联以提取绝对流体动力学半径,并且通过提取对于在多个流速下的多个“合并”的扩展物种的扩散比率来延伸对不均匀混合物的分析,并且将这些与已知流体动力学半径的物种的模拟结果对比。
本发明的装置和方法提供了一种可用于任何感兴趣的蛋白质物种的非扰乱检测和浓度测定的一般技术,无论溶液条件和等电点。
亲水性通道
在流体装置中使用亲水性通道作为标记研究的一部分进行了研究。流体装置被制备具有由上游第一和第二供应通道供应的会聚混合通道(见图15(d))。该装置中的通道是进行等离子体处理以在通道表面上产生亲水性硅烷醇基团的标准pdms通道。这些通道然后充满水,导致保持亲水性表面持续若干天。在刚结合的微流体装置上进行等离子体处理步骤。
第一供应通道供应液体流中的组分(胰岛素)以及第二供应通道提供液体流中的标记物(opa混合物)。允许来自第一和第二通道的流体在会聚混合通道的上游端在接合点处接触(见明场图像图15(b)。会聚的流体(见明场图像图15(b)和荧光图像15(e))稍后在检测区内进行分析(见明场图像图15(c)和荧光图像15(f)。
含胰岛素的流体含有10mg/ml在ph2的胰岛素。标记流体含有12mmopa、18mmbme、4%sds、以及200mm在ph10.5的碳酸盐。
图15(a)、(b)和(c)示出了虽然界面是清楚可见的(对于不同粘度的溶液预期的),胰岛素不会粘在处理的pdms通道上并且相反在下游溶解。即使在装置中使用高浓度胰岛素流体情况是这样。在这种情况下,预期一些不溶性蛋白质在该蛋白质越过其等电点时存在于层流界面处。不溶性蛋白质不会粘在pdms通道上并且相反在下游溶解。
在此的图像是与在图11和12(b)的图像中可见的不溶物质相对比。
图15(g)示出了在图15(d)的装置中产生了基本上稳定的流。这是由于堵塞的最小化和微泡形成的防止(由于更好的润湿)。
使用亲水性通道被认为消除了在本发明的方法中对单独的变性步骤的需要。在完全混合之前暂时不溶的蛋白质不会粘到通道壁并且稍后将在下游溶解。
电泳分离
电泳是用于分离核酸、肽和细胞的通用生物技术。凝胶电泳(其中分析物电荷与尺寸之比是在施加电场时在固体基质中经由阻滞评估的)是最常用的技术,虽然这不很好地适合于研究弱蛋白质缔合事件因为基质筛分本身的行为可能破坏相互作用。毛细管电泳(ce)涉及基于其有差异的电泳迁移率和在整个通道的电渗流的分析物的时间分离。在自由流电泳(ffe)中,样品在整个平面通道以压力驱动流移动,并且在施加电场后的分离垂直于流向。因为ffe是稳态技术,注射和分离是连续进行的。微流体自由流电泳(μffe),ffe的微流体小型化,具有通过减少焦耳热的效果提高分离分辨率,和与其他分离技术容易在线集成的优点(turgeon等人微型自由流电泳:理论与应用(microfree-flowelectrophoresis:theoryandapplications)394,187-198(2009)。urlhttp://dx.doi.org/10.1007/s00216-009-2656-5)。
μffe的限制之一历来是被集成到微流体装置内的电极,具有确保电极和导电水性介质之间的直接接触所必要的挑战性多步骤处理(kohlheyer等人芯片实验室(labonachip)6,374-380(2006);cheng等人芯片实验室(labonachip)11,2316-2318(2011)。最近,本发明的诸位发明人中的一些已经开发出具有并排放置在通道内的电极、并且适合于电泳的微流体装置(herling,t.w.等人)。
herling等人描述了在单个光刻步骤中将3维电极结合到微流体装置中,并且作者已经使用微流体装置来定量小分子的净溶剂化的电荷(herling,t.w.等人应用物理学快报(appliedphysicsletters)102,184102-4(2013))。在最初的工作中,然而,使用荧光染料以允许检测。这种分离生物分子的技术的应用要求使用被荧光标记的生物分子。外源性荧光标记的存在(其影响分子尺寸、电荷和相互作用)具有影响在观测中的过程的可能性。在实践中外源标记已被证明在电泳分离中是尤其有问题的。
如先前讨论的,在优选的实施例中,本发明提供了用于分离和后续标记流体装置内的组分(如蛋白质)的方法。分离后标记避免了上面讨论的问题。因此,没有影响分离之前和过程中的组分的行为,因为不存在标记物。
本发明的诸位发明人已经开发了连接到到检测步骤的电泳分离,该步骤包括分离后标记。图5(a)和(b)示出了可在本发明的方法中使用的根据本发明的装置的设计。
蛋白质和缓冲液被装入在图5(a)左侧显示的装置入口。缓冲液流体流在蛋白质流体流的任一侧提供。流体流接触大截面通道,并且结合的层状流体流进入小截面通道。本发明的诸位发明人中的一些先前已建立了在接合点处使用大截面通道最小化流体滞留。
电极被设置在小截面通道的任一侧并且是在组分跨过通道偏转时使用。大和小截面通道构成分离通道。在小截面通道的下游端,那里如果设置流分离器,该流分离器使结合的流体流的一部分转向。在图5(a)的装置中,流分离器使该缓冲液流的一部分转向。图5(b)是示出了流分离器的图5(a)的装置的特写图,其位于偏移小截面通道的纵向中心线。
允许在流分离器中的转向的流与标记混合物的流结合。标记混合物的流从储罐提供,如图5(a)中所示。在转向的流中的组分被标记并且随后进行分析。在流分离器下游的流通道构成检测区。
抵抗性是这样,当该装置是以退避模式通过施加压降操作时,缓冲液体积流速是蛋白质体积流速的十倍,使得蛋白质分布是尖的、“帽子”函数,被缓冲液的鞘侧面包围。蛋白质的“束”(是中央流体流)穿过分离通道,其在该通道的任一侧设置有电极。当跨过该通道施加电压时,蛋白质被偏转到缓冲液流,并且偏转程度与蛋白质的电荷与尺寸之比相关。偏转引导蛋白质朝向或远离流分离器,其使缓冲液流体流的一部分转向。在图5(a)和(b)中所示的流分离器被设计为转向离开分离通道的总流量的约10%。流被转向到检测区,其中蛋白质被标记并随后进行检测。
来自分离通道的未转向的流也被收集并与离开检测区的转向的流体流重新组合(如图5(a)中所示)。
转向的蛋白质以1:1的比例与潜在共价标记(lcl)溶液混合,(在此11mmopa,16mmbme,180mm碳酸盐,ph9.5)。溶液在通道(也称为等待环路)中混合若干秒,这既使蛋白质变性又定量标记了蛋白质。然后测量流体流的荧光强度。
装置在聚二甲基硅氧烷(pdms)中流延,其通过在固化前添加约0.2%w/v碳纳米粉末被着色黑色以减少荧光背景。黑色装置被对准,并且通过将荧光染料与蛋白质一起加载使用不干扰蛋白质标记物(例如opa标记物)吸收、激发或发射的激发和发射光谱证实光束偏转。在此,若丹明6g被选择作为示踪染料。在对准装置并且验证示踪染料的适当偏转后,改变所施加的电压并且测量在检测区的荧光强度。因为标记典型地是定量的(例如,使用opa),荧光强度与偏转蛋白质的浓度成正比。
该装置被用于分离和检测蛋白质bsa和溶菌酶,跨越在分离通道中一系列电压。图5(c)示出了对于bsa和溶菌酶的电压依赖性偏转分布。bsa具有4.7的等电点(ge等人生物材料科学杂志(聚合物版)(j.biomater.sci.polymered.)9,131-150(1998))并应在ph7下带负电荷。
溶菌酶具有11.4的等电点,并且应该在ph7下带正电荷(wetter等人生物化学杂志(journalofbiologicalchemistry)192,237-242(1951))。因此,bsa和溶菌酶在分离通道内在相反的方向上偏转,具有高斯状电压偏转分布。电压偏转分布的紧密性很可能可以通过增加流量或减少用于检测的在空间上转向的蛋白质的体积分数被进一步降低。
附加实验
支持本发明的附加实验提供如下。诸位发明人探讨了利用扩散技术来表征组分相互作用,如蛋白-蛋白相互作用。诸位发明人还制备了其流体装置的另外的实施例,并且使用这样的装置来分离和标记流体流中的组分。
附加批量标记测量
各种荧光团、化学计量和变性条件分别使用荧光光谱仪(瓦里安,caryeclipse)和荧光微板阅读仪(bmglabtech),在石英荧光比色皿(hellma),或半区非蛋白质结合微板(康宁(corning),产品#3881)来研究。
在附加工作中使用的定量标记混合物包括:12mmopa,18mmbme,和4%w/vsds在200mm碳酸盐缓冲液中,ph10.5。标记溶液在室温下避光,并在制备3天内使用,或冷冻并在制备14天内使用。这种标记溶液典型地与感兴趣的样品1:1v/v混合。
除非另有说明,蛋白质溶液在5mmhepes,ph7.0中制备,并且其浓度在nanodropuv-vis分光光度计上分光光度确定。
时间控制的荧光测量使用装配有注射器模块的clariostar微板阅读仪(bmglabtech)进行。测量是在“孔模式”下进行,意味着单独处理每个孔。注射器模块将50μl染料以430μl/s的速度注入到单个孔中,搅拌该板1s,并且然后在移动到下一个孔之前对于125s的持续时间每0.25s测量样品。每种样品和染料背景溶液一式三份制备。
附加微流体设计与制造
如前所述,使用autocad软件(欧特克公司(autodesk,inc.))设计微流体装置。然后得到乙酸盐二元掩膜(微光刻服务(microlithographyservices)),具有对应于微流体装置中的通道的透明区域,和对应于背景的黑色区域。制备了具有图24中所示的设计的装置。
微流体装置是使用标准软光刻技术制造的。制备了呈现待流延装置的正影像的硅主模。该装置的高度通过将所希望的厚度的负性环氧树脂光致抗蚀剂(microchem,产品#su-83025,取决于所希望的厚度)旋涂在硅晶片上设定。在此处描述的附加工作中使用的装置是25μm高。最终对应于负性装置通道的正特征通过用乙酸盐掩模阻挡光致抗蚀剂的一部分,用校准的uv光交联曝光区域,并且用丙二醇单甲醚乙酸酯(pgmea)显影剂(microchem)除去未交联的聚合物,根据制造商的说明产生。
微流体装置被流延在聚二甲基硅氧烷(pdms)中。pdms弹性体和固化剂(道康宁,产品#184)分别以10:1重量比混合。重要的是混合是充分的:人工搅拌至少5分钟对于最佳固化弹性体性能是重要的。如果黑色pdms被流延,这是通过将约1mg/ml的碳纳米粉末(西格码(sigma),产品#633100)添加到弹性体/固化剂中,并充分混合制备的。
该装置是通过在出口处的流体的抽取操作的。通道的尺寸因此被选择为基于液压和电气回路之间的类比控制通过该装置的流体流的相对速率。在如图24中所示的代表性的基于扩散的装置中,蛋白质溶液和缓冲液溶液在分离通道的上游端以1:1的体积比接触,并且蛋白质和缓冲溶液的接触流在分离通道中以25μl/小时的速度向下游行进。然后从组合流转向所得的流束(其含有从含有初始蛋白质流扩散最远的蛋白质)的三分之一。此转向的部分,它是缓冲液流的一部分(仅),然后与该流中的opa荧光标记混合物的流以1:1的体积比接触。体积比可以根据感兴趣的系统而改变。
基于图23中列出的动力学测量,荧光检测在opa荧光标记流和转向的流的初始接触3.1s后发生。
发现组分(如蛋白质),其在流内标记反应过程中越过它们的等电点,具有粘到流动装置的疏水性pdms上的倾向。如果pdms通道被制成更亲水的,则这个问题被消除(并且总体流量稳定性增加)。
为了实现这种增加的亲水性,使密封的pdms通道经受一个第二延伸的等离子体氧化步骤,以在通道表面上产生硅烷醇基团,如下面描述。
然而,氧化的黑色装置比氧化的透明装置更容易非特异性蛋白质吸附,可能是因为碳缺陷在表面的存在可能使硅烷醇层更容易破裂和疏水恢复。
因此,开发了“夹层式”装置,其中通道在透明pdms的薄层中流延,然后将其在顶部和侧面用黑色pdms覆盖以减少荧光背景。
因此,约2mm厚的透明pdms层被流延并且烘烤60min。透明pdms装置被切割并且可逆地结合,通道侧向下,到已用氮气清洗的皮氏培养皿。重要的是确保围绕透明pdms装置进行精确切割:发现沿着边缘的缺陷导致不良的结合并将允许黑色pdms渗到透明pdms下方并且进入通道特征
为了进一步减少渗漏,黑色pdms明智地是从透明装置制备的,已经允许该装置在室温下固化几小时以提高基底的粘度。黑色pdms然后可倒入可逆结合的透明装置的顶部,达约3mm的总添加高度。将夹心式装置烘烤75分钟。
烘烤后,混合装置容易从皮氏培养皿中移除,并且将装置开孔。使用0.75mm直径uni-core打孔器(harris)在入口和出口处打孔。在粘合之前,碎屑用“魔术贴”(scotch)和ipa超声移除。重要的是在粘合之前除去残留的ipa(其被吸收到pdms),所以将装置用氮气吹干,并且在结合步骤之前烘烤15分钟。
最初粘接涉及10s氧等离子体的产生。装置被密封后,将它们烘烤10分钟。以允许完全密封的形成。随后,将密封的装置在高功率下以500s氧等离子体产生再次进行氧化。该装置在氧化步骤(使用protex0.38mm内径,1.09mm外径管,连接到1ml塑料air-tite注射器)后立即用水充满,这防止在通道内的疏水性恢复。入口和出口用充水凝胶装载端头阻断。用这种处理,装置成功地抵制在结合和氧化后至少七天的非特异性蛋白质吸附。
微流体装置的使用
本发明的一个示例性装置在图24中示出,用于在基于扩散的分离方法中使用。将折叠的、未标记的、天然蛋白质分子和缓冲液的相等部分装入微流体装置中。当流体被限制在微米尺度时,这些流是层流的,而不是对流的,使得当组分和缓冲液的流束与微流体通道接触时,组分跨过通道的空间分布在任何限定的停留时间后是完全通过分析物扩散系数确定的。
关键地,一旦系统已经达到了良好定义的起始状态就开始测量。由于微流体方案中没有湍流混合,在t0,所有rh的组分具有相同的初始分布,占据扩散通道的一半宽度。这种情况通过对于模拟的0.5和10nm物种的横向浓度梯度的等效性在图24中示出。然后允许该系统在扩散发生时演变限定的时间段。在随时间td-t0扩散后,较小的0.5nm物种已比10nm物种扩散更远。
对于在稳态下操作的微流体系统观察到的与流向垂直的空间分离-而不是平行于流向的暂时时间分离,允许空间分布的一小部分连续转向到下游模块,没有分辨率的损失。
整个装置内的相对流体流通过改变沿可能路径的压差精确地设定,这是通过用通道尺寸控制流体动力学阻力实现的。以这种方式,在时间td-t0(矩形)扩散通道宽度的至少六分之一的分布的一小部分是在时间td指向标记模块。在这个位置,通过在同向流动接合点i2处引入标记溶液改变条件,以允许所有反应蛋白基团的定量修饰。
因为潜在荧光团本身不是荧光的,无需纯化步骤。如果蛋白质序列,以及因此反应性基团的数目是已知的,荧光强度的测量允许用于测定绝对蛋白质浓度。
缓冲液和装载样品在使用之前最初通过0.22μm滤器(millipore)过滤,以消除可能堵塞装置的微粒物质。装置通过用适当的天然缓冲液从出口填充来装载。
总体上,使用1ml汉密尔顿(hamilton)玻璃注射器或1ml塑料air-tite注射器(通过27号计量注射针连接到portex管)来控制流体通过装置的流动。
在这些实验中使用的流速下玻璃和塑料注射器的性能之间没有注意到差异。在入口和注射器同时施加压力以除去在装载过程中形成的任何气泡,并且在装置入口用凝胶装载端头引入试剂。
试剂装载量在10和200μl之间变化,取决于实验。应注意的是可以在本发明的方法中使用较小的体积。
如与前面所描述的实验一样,用nemesys注射泵通过装置抽取流体。为了在装载步骤过程中最初通过装置汲取试剂并且最小化任何入口错流的影响,以300μl/小时的流速最初抽取20μl流体。对于在实验中使用的基于扩散的装置,使用在分离通道中的25μl/小时的流速,其对应于在该装置的出口处的33.3μl/小时的抽取速率。允许流速在图像采集开始之前平衡至少18分钟。
明场和荧光图像是使用zeissaxioobserver显微镜获得的,该显微镜配备有coolsnapmyoccd照相机(photometrics)、365nmcaimoptoled(photometrics),和用于荧光图像的chromo49000dapi过滤器(photometrics)。使用了2.5x、5x、10x和20x物镜。在附加工作中使用在10ms和10s之间的曝光时间,并且总体上在每个采集过程中在10和60个图像之间进行平均。
当荧光信号是低的时,使用em增益,或合并相邻像素。对于每一组测量和成像设置,拍摄至少一个染料背景图像以将未反应的染料的最小荧光考虑在内。还获得了平场背景图像。在黑暗环境中进行测量,并且将在分析过程中的温度保持在25℃。
图像分析及配件
常规地获得了接合点(如分离通道的上游区域)的图像,通道(如检测通道,其中标记流与转向的流接触)和流分离器。在这些图像显示由于堵塞或其他异常改变的流分布时,丢弃在该检测区获得的图像。
在imagej进行基本图像分析。从获得的每个图像中减去平场背景图像。简略地,在停留时间标记的上游直接定义感兴趣的区域(丢弃装置壁附近的区域,在那里由于无滑动边界条件流速显著降低)。计算在这个区域内的平均荧光,并从该通道的正上方和下方的芯片的平面场区域的此平均荧光中减去,这降低了照明源强度随时间的变化,和样品吸附到pdms的影响。
样品流体动力学半径是通过将实验强度比与对于已知尺寸的参考颗粒模拟的那些相比较计算的。因此,对于每一个样品,计算以下比率:
其中φ是比较在后续分析中使用的扩散和均匀分布的样品的强度比,γ1是在检测区中的背景校正的荧光强度,当样品被装载到一个装置入口时被观察到,γ2是在检测区中的背景校正的荧光强度,当样品被装载到装置的两个入口时被观察到,并且γd是opa标记混合物的背景校正的强度。φ被用于基于从对于基函数观察到的插值确定每一个样品的流体动力学半径。
芯片上吸收
批量吸收测量在nanodrop分光光度计上使用标准蛋白质a280设置进行,虽然在使用瓦里安(varian)uv/vis分光光度计时获得了类似的结果。这在下面的批量吸收部分中描述。
芯片上uv吸收测量使用商业环烯烃共聚物芯片(thinxxs,通道截面320×320μm)和actipixd100uv区成像检测器(paraytec)进行。总装置厚度为1.7mm,并且在黑暗环境中进行所有测量。
允许含有150μmbsa的溶液流动通过该装置的一个入口并且milli-q水以50μl/h的速率通过另一个以各自形成界面。该通道通过由仪器的脉冲氙灯(在280nm下过滤的带通)的8个连续信号突发以突发之间10ms的延迟照亮。随后的光强度在100ms的时间跨度积分。在只用水填充的通道所进行的测量下的背景校正给出了约120的信噪比。这导致对于几μm的装置设置的检测限。
应当注意的是,在熔凝二氧化硅毛细管中,已经检测到下降到大约100nmbsa的浓度。
批量吸收
将对于批量吸收测量观察到的灵敏度与本案的方法进行对比。
发现要求大约600nm的蛋白质来通过批量吸收精确地确定蛋白质浓度(见图20),而低于nm的蛋白质浓度可以使用本案的方法精确地批量确定,其中它们使用opa-标记程序。
通过从浓缩的储备溶液稀释制备了许多不同浓度的bsa样品,其浓度已经分光光度确定。a280被描绘为如图20中所示的蛋白质浓度(nm)的函数。在标记被放置在正方形中时,这表明该样品比缓冲液空白吸收较少的光。需要约600nmbsa从a280精确地确定蛋白质浓度。
非特异性蛋白质吸附和流动稳定性
本发明的微流体装置典型地在聚二甲基硅氧烷(pdms)中制造。pdms软光刻技术的优点是公认的,与其他光刻方法比较最值得注意的是快速成型,成本低,且高通量。然而,在低于组分的等电点的ph下组分(如蛋白质)被输送通过流体装置时出现了问题。
在本案的优选的标记步骤中,标记混合物将流体流的碱度增加至约ph10.5。
当组分如蛋白质在芯片上越过其等电点(iep)时,使得存在于含有组分的流(例如可能存在于转向的流中)和标记流之间的层流界面处的组分暂时不带电荷,并且该组分被视为粘附到pdms通道表面。这样的粘附阻断了微流体接合点,导致流中断。这可能导致检测区中所产生的荧光随时间的显著变化。这使得所记录结果的定量解释困难并且有时是不可能的。
为了解决这一问题有帮助的是在已结合到玻璃表面的pdms装置上进行附加等离子体氧化步骤。这个附加的氧化处理在pdms通道的暴露表面上形成亲水性的、玻璃状硅氧烷层,这降低了非特异性组分吸附并且大大改善了流动稳定性。
然而,当通道在黑色pdms中流延时这些有益的特性丢失,最有可能是由于在碳缺陷在硅氧烷表面的存在下的加速的疏水性恢复。在这项工作中诸位发明人已开发了夹层式装置,这些装置结合了有效的通道氧化和降低的荧光背景的益处。
图21示出了对于在流体装置中越过其iep的牛胰岛素随时间(s)的归一化荧光(au)的变化,其中(a)是根据本发明的实施例的标准pdms流体装置;(b)是已经经受附加的等离子体处理的标准pdms流体装置;以及(c)是已经经受附加的等离子体处理的标准黑色pdms流体装置。
在标准装置(不经受附加等离子体处理的装置)中在流体通道中观察到显著的聚集沉积。由此得出存在归一化的荧光信号随时间的显著变化(如在图21(a)中看出)。pdms表面的延伸的等离子体氧化产生了抵抗蛋白质吸附的富含硅烷醇的亲水层。当透明pdms通道以这种方式被氧化时,它们抵抗在层流界面处的蛋白质沉积,这允许显著改善的流动稳定性(如在图21(b)中看到的)。然而,当黑色通道被氧化时,对非特异性蛋白质吸附的一些阻力丢失,从而导致与透明装置相比流动稳定性的轻微下降(比较图21(c)与图21(b))。为了解决这个问题,诸位发明人已经使用了混合“夹层式”装置,其中通道由透明的pdms形成以促进有效且持久的氧化,而在顶部和侧面覆盖的黑色pdms减少了荧光背景。
rh确定
在本案的流动系统中,该系统在稳态下工作。在检测区下游的组分浓度的测量显示用于标记被转向的组分的总浓度。虽然空间分布在分析数据中不是直接可视化的,当进行附加测量时组分rh是可获得的。
图22(a)示出了在时刻td已知浓度的组分的均匀参考分布(线,在1.0颗粒浓度)。在实践中,这种分布可以容易地通过在第一和第二流体流中都提供组分来实现。
每个rh的物种(图22(a)中色度指示的)具有扩散相对用于标记转向的均匀分布物种的特征部分(转向步骤将已经扩散最远的物种转向至第二流)。因为标记基本上是定量的,很容易通过在检测区域对于这些物种对比观察到的荧光强度进行评估(图22(b))。如预期的,较小的物种占优势,因为这些物种在分离步骤过程中扩散最远。
在图22(a)和(b)中的数据是在图8和9中的数据的变化并且相一致。
为了测试方法和装置使用扩散分离和标记来尺寸排列组分的能力,开发并测试了尺寸阶梯。该阶梯包括在分子量(mw)的三个数量级内变化的生物分子。该尺寸阶梯附加地包括在二级和三级结构变化的蛋白质,未折叠的以及折叠的蛋白质和蛋白质复合物。
图22(c)示出了如通过分析超离心(auc)和脉冲场梯度nmr(pfg-nmr)确定的对于不同生物分子报道的文献流体动力学半径值和来自本案的基于扩散的方法的实验得出的值之间的对比。特别地,pfg-nmr被用于具有低消光系数的低mw重量物种,并且auc被用于较高mw重量物种。在可能时报道了这两个值。重要地,auc和pfg-nmr均不适合研究的整个分子量范围。
相比之下,在所研究的整个mw范围内使用本案的方法获得的流体动力学半径与使用auc和nmr技术的复合获得的那些非常相似。
在高分子量范围内的不确定性可以通过标记分布的不同部分,例如通过收集具有较高浓度的较高分子量物种的第二流的不同部分,或通过收集第一流的部分(其将在较低分子量物种中耗尽)来减少。
相对于预测的“自然”扩散流体动力学的值进行了基于扩散的rh值的进一步比较:其是可以含有指示分子量的蛋白质的最小球体(r最小)的流体动力学半径。该对比在图22(d)中示出。
r最小预测的准确度随mw增加而减小,可能反映了具有较长序列的蛋白质可获得的非球形构造。
该尺寸阶梯包括可以是对于使用传统尺寸方法表征具有挑战性的蛋白质和蛋白质复合物。蛋白质α-突触核蛋白在帕金森症中起着重要作用。因为α-突触核蛋白的天然未折叠结构是不紧凑的,计算的r最小,以及从auc测量获得的rh比使用pfg-nmr或本案的扩散方法获得的那些显著更小。
本发明的基于扩散的方法被用于分析蛋白质的非均匀混合物。制备了含有zn2+游离牛胰岛素的水性样品,其中单体和二聚体形式处于平衡。从扩散方法测定的rh值是1.64±0.16nm,这反映了在样品中存在的单体和二聚体的比例。
组分相互作用
本案的方法可用于研究组分(如蛋白质)的组装,具有组分浓度的变化。例如,扩散分离技术可用于研究在溶液中的物种的流体动力学半径的变化。
复合流体动力学半径可以基于复合物种的相对丰度,以及纯组分的半径来计算。在一般情况下,当对单个组分进行分析时:
rh=fs(φ)
其中,rh是所观察到的流体动力学半径,φ实验观察到的对于扩散相对于均匀分布的颗粒的强度比,以及fs是基于比较φ与对于基函数模拟的插入了未知样品的尺寸的尺寸函数,如在此所述的和相对于wo2014/064438。在非均匀混合物的情况下:
强度比的线性组合,φi,和比例,n种混合物组分的每个i的pi给出rh。应注意的是,pi描述了组分i有助于总伯胺浓度,而不是总蛋白浓度的伯胺的比例。
进行了一系列实验以探讨复合rh是否可用于提取结合常数,和纯混合物组分的尺寸,用于蛋白质-蛋白质相互作用的定量表征。将来自本发明的扩散分离方法的结果与通过动态光散射(通常使用的扩散尺寸技术)获得的结果进行比较。
对β-乳球蛋白的低聚进行了研究。虽然β-乳球蛋白是通常使用的生物物理模型蛋白质,在文献中已经报道了超过一个数量级变化的平衡常数,且关于在生理浓度下的低聚是否仅提供二聚体,或四聚体和八聚体是否也存在于群体中存在分歧。
在中性ph和5mm离子强度下制备了一系列β-乳球蛋白溶液,具有范围从2至40μm的β-乳球蛋白的浓度。将溶液通过dls和在此所描述的扩散分离方法进行分析。低聚结果呈现在图16中,其中示出了对于在溶液中不同浓度的β-乳球蛋白(μm)所计算的流体动力学半径rh(nm)用于dls实验(a)和扩散实验(b)。数据以zave值呈现。
在dls实验中,只有在最高蛋白浓度下想起对于获得的蛋白质二聚体所报告的值的尺寸。然而,仍然存在数据的显著变化并且仍然存在数据的显著变化。通过dls不可能观察群体中的蛋白质单体。
由本案的扩散方法获得的复合rh示于图16(b)中。流体动力学半径在所研究的浓度范围内增加1.83和2.60nm,对应于1.4的尺寸增加。这与二聚化事件是一致的。为了进一步阐明低聚机制,将数据拟合以获得对于纯单体和二聚体的φi和pi,如上所述。传统上,在均二聚体化事件中:
[m]+2[d]=ct
其中m和d分别是单体和二聚体的摩尔浓度,
ct是存在于混合物中的所有物种的总浓度,并且kd是二聚化常数。解组合的二次方程得到单体浓度,并考虑单体贡献的伯胺的比例的化学计量,mpa
等效地,二聚体伯胺浓度很容易被确定为:
[dpa]=[ct]-[mpa]
得到相关pi拟合参数。以这种方式拟合数据,得到4.1μm的解离常数,连同对于纯单体和二聚体分别为1.34和3.21nm的推测尺寸。
使用本案的方法,观察到所测量的流体动力学比所研究的浓度范围(从约1μm至40μm)增加了1.42nm。呈现的半径是复合值,将所有混合物组分考虑在内。
为了促进蛋白质-蛋白质相互作用的定量分析,诸位发明人以前已经开发了方法来从这样的复合数据提取平衡常数和纯混合物组分的尺寸,例如如在此所描述的和如wo2014/064438中所描述的。
如以上所指出,在研究中获得的数据很好地拟合到二聚化模型,其揭示了4.1μm的解离常数,对矛盾的β-乳球蛋白二聚化事件的文献报告有新的阐述。
因此,可以使用本发明的方法来识别混合物中的单体和组装的组分。
蛋白质三元混合物的分离
自由流电泳分离在分离物种的复杂混合物的组分中比扩散分离更有效。在扩散分离情况下,低分子量物种可从高分子量物种分离,但高分子量物种用低分子量物种的背景洗脱。相比之下,在自由流电泳分离情况下,通过改变装置的几何形状,可以得到高的分辨率,并且纯组分可以被分离(也参见herling等人)。
流体装置结合电泳分离通道被制备,具有下游流分离器和检测区。提供该流分离器和该检测区之间的流体连接的是标记通道。该装置的示意图在图17(a)中示出。该装置是对图5和14的装置的改变。该装置在自由流电泳分离和转向后包括稳态分离后标记步骤。该装置是使用标准pdms软光刻技术制备的,如在装置制造部分如上所述的。
组分(如蛋白质)的明确定义的“束”是通过在组分流的任一侧提供缓冲液流建立的。建立层流。分离通道内的组分流的宽度是基于改变组分和缓冲液流的相对流速可调的。
当没有跨过分离通道(跨过层流的宽度)施加电压时,该组分不被转向到相邻缓冲液流中。因此,在不存在施加场时,组分流内的组分不被转向到标记通道和检测区。设置流分离器以在分离通道的下游端转向缓冲液流的一部分(仅)。在分离通道的下游端的组分流中存在的组分被简单地收集在废物通道中。
当跨过分离通道施加场时,组分可以从组分流被偏转到缓冲液流,并且这两个侧缓冲液流的哪一个将取决于组分的电荷和所施加的场的方向。被偏转到缓冲液流的组分可以由流分离器转向,并且转向的流被带到下游。在下游区域中转向的组分可以被标记,如荧光标记,用于在检测区域中的检测。
流分离装置使用五种不同的蛋白质溶液进行测试。制备了含有bsa、β-乳球蛋白和溶菌酶(1.0mg/ml)之一的三种蛋白质溶液,还制备了含有bsa和溶菌酶的二元溶液(1.0mg/ml的总蛋白质浓度;0.5mg/ml对于每种蛋白质),以及含有bsa、β-lac和溶菌酶的三元溶液(1.0mg/ml的总蛋白质浓度;0.33mg/ml对于每种蛋白质)。含有溶菌酶的溶液另外包括1%v/v的吐温表面活性剂,以便最小化在标记步骤过程中溶菌酶的沉淀。
每种蛋白质溶液的流在流体装置中建立,并允许蛋白质流沿着分离通道通过。改变跨过通道施加的场,并且监测每种蛋白质响应于所施加的场的偏转。偏转到缓冲液流中的蛋白质可以通过转向在分离通道的下游端的缓冲液流的一部分来收集。转向的缓冲液流,含有蛋白质,然后与标记流接触,以便荧光标记蛋白质用于检测。标记流包括变性剂和潜在标记opa,将其如在此所述使用。标记后,蛋白质通过荧光光谱法检测。所记录的信号的强度与从在分离通道的下游端的缓冲液流的一部分转向的蛋白质的浓度成正比。
图17(b)示出了对于以上描述的五种溶液中的每一种归一化的荧光强度(au)随跨过分离通道施加的场(v)的变化而变化。
对于含有bsa的流,当跨过分离通道施加的场是大约4.0v时,在检测区中记录的荧光强度是处于最大值。
当跨过分离通道施加的场是大约6.0v时,对于含有β-乳球蛋白的流的荧光强度是处于最大值。当跨过分离通道施加的场是大约-6.0v时,对于含有溶菌酶的流的荧光强度是处于最大值。偏转分布是电压依赖性的并且对应于基于在实验ph值(对于每种溶液ph7.0)下蛋白质的电荷的蛋白质的预期偏转。
跨过分离通道的特定电压的施加可用于优先偏转多组分流中的感兴趣的组分。以这种方式,一种组分可以从多组分混合物中的其他组分中分离出。上述二元和三元混合物被用于验证一种组分与一种或多种其他组分的自由流电泳分离和标记。在二元和三元混合物中观察到特征峰,其对应于对于单独的蛋白质所观察到的峰。
特别感兴趣的是bsa和β-乳球蛋白之间的表观分辨率,其具有相似的等电点。
对于三元样品接近零电压所观察到的宽峰能够反射从带相反电荷的蛋白质之间静电相互作用产生的低聚物的分布。目前进一步的工作正在进行以对此进行研究。
定量标记:动力学
本发明的诸位发明人已经研究了多种策略用于实现定量标记。因为在opa-标记反应过程中形成的荧光团缺乏化学稳定性,像质谱等技术不理想地适合于评估组分标记的程度。
设计了其中定量标记可以在限定的时间在混合后直接进行评估的试验。蛋白质被用作测试组分。对于具有不同的二级和三级结构的很好地表征的蛋白质,将荧光强度与反应性基团的浓度(蛋白质浓度×在蛋白质序列中的伯胺的数目)进行比较。
参考组包括牛血清白蛋白(bsa),在ph2和ph7的β-乳球蛋白(β-lac),溶菌酶(lys),钙调蛋白(cam)和肌球蛋白激酶肽(p7)。参考组包括在标记反应过程中越过、或接近它们的等电点的蛋白质。
将对于该组中的蛋白质的标记反应与对于游离甘氨酸和赖氨酸的标记反应进行比较,其具有与在标记反应过程中改性的那些类似的反应性伯胺基团,但它们是完全溶剂可获得的(参见图23(a))。
发现了当荧光强度在混合120s后批量测量时,组合的变性策略-其包括加入4%十二烷基硫酸钠(sds)、过量的bme和高碱性反应混合物-得到伯胺浓度和荧光强度之间的线性关系(图23(b))。
该数据显示s形特征,然而,对于分别具有低和高伯胺浓度的混合物具有高于和低于预期的荧光强度。此外在甘氨酸和赖氨酸的标记效率中存在系统差异,具有对于在这些范围内的单独的蛋白质可见的趋势。
由于对于opa-改性的胺报告的低化学稳定性,推测这些效果可能部分地起源于动力学。产生最大荧光强度所需的时间在微流体混合装置内测量,如图23(c)中所示,并观察到标记反应在3s内达到完成。在稍后的时间点,荧光强度总体上以复合基底和浓度依赖的方式降低。
由于所报告的在产生荧光的opa-标记反应中形成的取代的异吲哚的化学稳定性的缺乏,我们详细研究了形成的动力学并且研究了荧光物种的降解。
在3s时间点构建了类似的定量标记评估,旨在测量在降解机理产生显著效果前的荧光强度(见图23(d))。关键地,当在反应完成后迅速测量荧光强度时,序列依赖性变化崩溃,并且线性在覆盖四个数量级的伯胺浓度范围内延伸,使能测量低于nm的蛋白浓度。对于参考,通过批量吸收测量常规可获得的浓度范围用灰色矩形突出。考虑到微流体系统的低路径长度特征,该检测限在本发明中使用的装置中进行了验证,其结合了3s的反应环路。
应指出bsa的浓度可以从在3.75nm和15um之间的值的荧光强度定量确定。鉴于其中荧光强度在微流体装置中定量的检测区的部分的体积是仅2.25nl,此结果表明小于9阿摩尔的bsa可在流动装置上定量。
将不同浓度的bsa、β-lac和溶菌酶与opa标记混合物混合,并且随时间(s)测量平均荧光强度(au)。结果在图18中示出。对于每种蛋白质,看到平均荧光强度随时间降低,具有在60s内平均荧光强度的明显下降。
观察到荧光强度随时间的轻微下降,可能是由于所形成的异吲哚缺乏化学稳定性。另外,对于低蛋白质浓度,观察到约40-60s的荧光强度的增加,可能是由于可能在强制条件下可操作的竞争性途径。如在本案中讨论的,在蛋白质与标记物反应后迅速测量荧光强度最小化了这些影响。
动态范围随时间的减小通过检查平均荧光强度在较长的时间范围内的变化被进一步说明。因此,平均荧光强度在从标记反应开始30分钟后进行测量。在该实验中,允许染料和蛋白质溶液在混合后在室温下静置。结果在图19中示出,其中平均荧光强度(au)被示为在溶液中的伯胺浓度(nm)的函数。将数据的双对数拟合为方程f(x)=0.99x,显示了0.67的r2。
在低蛋白质浓度下从线性度的偏离在较长的反应时间之后是更显著的。对于在30分钟内的荧光测量需要超过1μm的伯胺浓度来从荧光强度准确测定蛋白质浓度。在低于1μm的伯胺浓度和在非常高的胺浓度下,存在所记录的平均荧光强度与胺浓度的线性关系的偏差。据认为从竞争性反应途径产生的这种偏差在此更显著。
根据这些结果,诸位发明人现在认识到在组分(如蛋白质)被荧光标记后不久测量荧光强度是有益的。
参考文献
在本说明书中提及的所有文献均通过引用整体并入本文中。
ahmadcan生物化学与细胞生物学杂志(j.biochem.cellbiol.)63,1058-1063(1985)
almgren等人胶体与界面科学杂志(journalofcolloidandinterfacescience)202,222-231(1998)
benson等人美国科学院院报(pnas)72,619-622(1975)
biancalana等人生物化学与生物物理学学报(biochimicaetbiophysicaacta)1804,1405-1412(2010)
brandts等人美国化学会志(j.am.chem.soc.)89,4826-4838(1967)
brody等人传感器和致动器a:物理(sensorsandactuatorsa:physical),58(1):13-18,1997
cheng等人芯片实验室(labonachip)11,2316-2318(2011)
cohen等人分子生物学杂志(journalofmolecularbiology)421,160-171(2012)
flockhart等人胶体科学杂志(journalofcolloidscience)12,557-565(1957)
garciaalvarez-coque等人分析生物化学(anal.biochem.)178,1-7(1989)
gb1320127.2
ge等人生物材料科学杂志(聚合物版)(j.biomater.sci.polymered.)9,131-150(1998)
hatch等人自然生物技术(naturebiotechnology),19(5):461-465,2001
hellstrand等人acs化学神经系统学(acschemicalneuroscience),1,13-18(2010)
herling,t.w.等人应用物理学快报(appliedphysicsletters)102,184102-4(2013)
hirota等人蛋白质科学(proteinscience)6,416-421(1997)
ivanova等人美国科学院院报(pnas)2009,106(45),18990-18995
jacobs等人分析生物化学(analyticalbiochemistry)1986,156,334
jacobson等人分析化学(anal.chem.)1994,66,4127
jacobson等人分析化学(anal.chem.)1994,66,3472
jimenez等人美国科学院院报(pnas)99,9196-9201(2002)
kamholz等人生物物理学杂志(biophysicaljournal),80(4):1967-1972,2001
kang等人芯片实验室(labonachip)8,176-178(2008)
kim,p.等人生物芯片杂志(biochipjournal)2,1-11(2008)
knowles等人科学(science)326,1533-1537(2009)
kohlheyer等人芯片实验室(labonachip)6,374-380(2006)
lauga等人urlhttp://arxiv.org/abs/cond-mat/0501557
levine等人蛋白质科学(proteinscience)2,404-410(1993)
lee等人芯片实验室(labonachip)9,479-482(2009)
liu等人分析化学(anal.chem.)2000,72,4608
mok等人方法(methods)54,67-75(2011)
monahan等人分析化学(analyticalchemistry)73,3193-3197(2001)
nakamura等人分析快报(analyticalletters)1982,15,1393-1410
nettleton,e.j.等人生物物理学杂志(biophysicaljournal)79,1053-1065(2000)
oguriet等人色谱杂志(journalofchromatography)a787,253-260(1997)
otzen生物化学和生物物理学学报(biochim.biophys.acta)1814,562-591(2011)
otzen生物物理学杂志(biophys.j.)83,2219-2230(2002)
pct/gb2013/052757(wo2014/064438)
powers等人生物聚合物(biopolymers)33,92-932(1993)
rida等人分析化学(analyticalchemistry)76,6239-6246(2004)
roth等人分析化学(analyticalchemistry)43,880{882(1971)
roth等人色谱杂志(journalofchromatography)83,353-356,(1973)
saito等人分析化学(analyticalchemistry)1994,66,134-138
schuck分析生物化学(anal.biochem.)320,104-124(2003)
squires等人现代物理评论(reviewsofmodernphysics)77,977-1026(2005)
sternson等人分析生物化学(anal.biochem.)144,233-246(1985)
stone等人流体力学年鉴(annualreviewoffluidmechanics)36,381-411(2004)
tan等人生物微流体(biomicrofluidics)4,032204(2010)
turgeon等人微型自由流电泳:理论与应用(microfree-flowelectrophoresis:theoryandapplications)394,187-198(2009)。
urlhttp://dx.doi.org/10.1007/s00216-009-2656-5
us2002/0186263
us2006/0263903
us2010/0032349
us2011/264380
us2012/0135507
walsh等人febs杂志(febsjournal),276,1266-1281(2009)
waugh美国化学会志(j.am.chem.soc.)68,247-250(1946)
wetter等人生物化学杂志(journalofbiologicalchemistry)192,237-242(1951)
whitesides自然(nature)442,368-373(2006)
whittingham等人分子生物学杂志(journalofmolecularbiology)318,479-490(2002)
wong等人美国化学会志(j.am.chem.soc.)107,6421-6422(1985)
yoshimura等人分析生物化学(anal.biochem.)164,132-137(1987)
zawieja等人分析生物化学(analyticalbiochemistry)142,182-188(1984)
- 还没有人留言评论。精彩留言会获得点赞!