快速制备标记的葡糖基胺和分析生产所述标记的葡糖基胺的糖基化生物分子的方法与流程
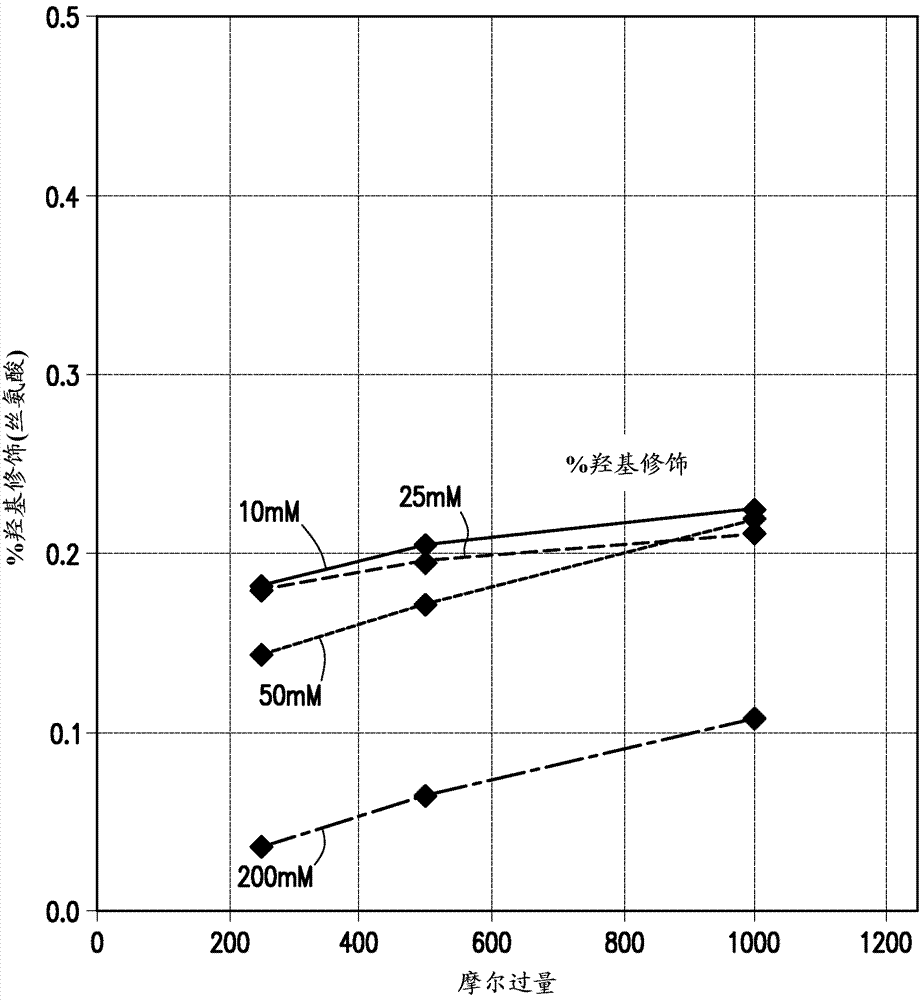
相关申请的交叉参照本申请要求2014年10月30日提交的美国临时申请号62/072,747和2015年1月26日提交的美国临时申请号62/107,994的优先权,其全文通过引用结合到本文中。关于联邦政府赞助的研究或开发的声明无。联合研究协议的当事人名称无。背景分析来自生物源的化合物的方法常常包括衍生化步骤,以在色谱分离后引进有利于检测的荧光团。虽然存在不同的衍生化策略,但长处理时间是一个有效地采用一旦获得的分析数据的严重因素。最近已开发出减少前置时间(leadtime)的试剂和相关方法,特别是关于具有胺或氨基的化合物的分析。然而伯胺和羟基之间的期望的反应选择性常常不能实现。进而,标记化合物的得率则不能优化。此外,现有技术方法频繁地生成“过度标记的”化合物。而且,标记的(labeled)或加标签的(tagged)化合物在高有机溶剂中的溶解性可能是低的,这阻碍下游固相提取程序(“spe”),尤其是基于亲水相互作用色谱的那些。同样,现有技术分析通常需要化合物从生物源分离和/或在衍生化之前经历洗涤步骤,这产生额外的步骤并减慢分析程序。因此,存在对分析化合物的方法的需求,其中衍生化步骤以高得率生产被选择性地标记的加标签化合物,且不引起生物样品的降解或过度-标记,以在标记化合物的随后的分析期间提供高的分辨率。发明概述一种分析糖基化生物分子的方法,其包括以下步骤:(a)生产去糖基化混合物,其中糖基化生物分子已通过天然或合成酶或化学技术去糖基化;(b)提供包含在极性非质子、非-亲核有机溶剂中的标记试剂的试剂溶液;(c)在反应混合物中,以约2.5:约1的体积比混合去糖基化混合物与试剂溶液;和(d)检测衍生的葡糖基胺。反应混合物含有相对于可修饰的胺的范围从约10-约2000的量的摩尔过量的标记试剂,并可包括释放的葡糖基胺、蛋白质性胺和衍生的葡糖基胺。所述步骤可有目的地进行,而不损耗蛋白质物质。然后衍生的葡糖基胺可用在线或离线spe和/或其它分离方法从反应混合物分离,或不从反应混合物分离。任选地,猝灭溶液可加入到反应混合物中,以致反应混合物的ph移至高于10。衍生的葡糖基胺的得率可以是反应混合物的约80-约100摩尔%的量。衍生的葡糖基胺然后从反应混合物分离并通过色谱检测、荧光检测、质谱(“ms”)或紫外光(“uv”)检测和/或其组合检测。在一些实施方案中,所述方法还可包括使糖基化生物分子与酶接触以生产去糖基化混合物的步骤,或可通过其它酶或化学技术去糖基化。在另一方面,葡糖基胺的快速衍生的方法也在本文中描述。在一些实施方案中,葡糖基胺的快速衍生的方法包括以下步骤:(1)提供生物样品;(2)合并生物样品与肽n-糖苷酶f以生产去糖基化混合物;(3)提供包含与无水二甲基甲酰胺(dmf)合并的标记试剂的试剂溶液;和(4)混合去糖基化混合物与试剂溶液,以生产包含标记试剂、释放的葡糖基胺、蛋白质性胺和衍生的葡糖基胺的反应混合物。所述反应混合物可具有相对于可修饰胺的约10-约1000的量的摩尔过量的标记试剂。对于一些实施方案,摩尔过量的其它范围也在本文中描述。在所述方法中,有机溶剂在反应混合物中的浓度可以是约0-约50%。在一些实施方案中,有机溶剂在反应混合物中的范围可以是约10-约40%。在一些示例性实施方案中,有机溶剂在反应混合物中的范围可以是约20%-约30%体积。去糖基化混合物与试剂溶液以约9:1-约1:9的体积比混合。在示例性实施方案中,去糖基化混合物与试剂溶液以约2.5:约1的体积比混合,以产生具有约25-约30%的试剂溶液的反应混合物。在一些实施方案中,试剂溶液在反应混合物中的量是28.6%。试剂溶液可以是处于维持在约环境温度或少于环境温度-约4℃的温度下。缓冲溶液可加入去糖基化混合物中。缓冲溶液可以是磷酸钠或hepes。或者,生物样品可以在缓冲溶液,例如hepes中去糖基化,以用更少的步骤促进随后的衍生化反应。任选地,猝灭溶液可加入到反应混合物中。猝灭溶液可包含乙二胺。乙二胺:水:乙腈的比例可以是约5:约5:约90体积。然后可在去糖基化混合物与试剂溶液混合后约2-约10分钟,将猝灭溶液加入到反应混合物中。反应混合物的ph可移至约大于10。一般地,为制备供分析的糖基化生物分子,它可以被合成地或者天然地去糖基化。更特别地,为制备标记的n-聚糖,生物样品可具有糖基化生物分子例如糖蛋白,其可以用肽n-糖苷酶f(pngasef)去糖基化,生产去糖基化混合物。标记试剂可以在无水二甲基甲酰胺(dmf)和/或其它极性非质子非-亲核有机溶剂中合并,以生产试剂溶液。去糖基化混合物和试剂溶液随后以约2.5:约1的体积比(2.5:1v/v/v)混合,以生产反应混合物,且葡糖基胺可以用标记试剂快速地标记。衍生的葡糖基胺可以经历色谱分析、质谱、紫外光和/或荧光检测。在此描述的快速标记葡糖基胺的方法可包括可选择类型的分离和检测方法。分离技术可包括,但不限于,亲水相互作用色谱(“hilic”)、固相提取(“spe”)、毛细管电泳、高ph阴离子交换色谱或反相液相色谱。检测方法可包括色谱检测例如高压液相色谱(“hplc”)、超高效液相色谱(uhplc)、超临界流体色谱、紫外光(“uv”)检测、荧光检测、基质辅助激光解吸离子化质谱、电喷雾离子化质谱和/或脉冲安培检测。附图简述图1是显示在此描述的方法的步骤的流程图。图2是显示温度对氨基-标记的基团的得率和对胺和缓激肽的羟基之间的反应选择性的影响的图。图3a和3b各自是显示有机溶剂(分别为dmso和dmf)和有机溶剂浓度对氨基-标记的基团的得率和对胺和缓激肽的羟基之间的反应选择性的影响的图。图4a和4b是显示缓冲液(分别为200mm硼酸钠、200mm磷酸钠)对氨基-标记的基团的得率的影响的图。图4c显示当200mm硼酸钠和200mm磷酸钠缓冲液用于反应混合物中时,在羟基上修饰的葡糖基胺的%。图5a和5b是显示缓冲液浓度和离子强度对氨基-标记和羟基的得率和对胺和缓激肽的羟基之间的反应选择性的影响的图。图5a显示缓冲液浓度和离子强度对缓激肽的氨基-标记的基团的得率的影响。图5b显示缓冲液浓度和离子强度对缓激肽的羟基得率的影响。图6是显示反应时间对氨基-标记的基团的得率和对胺和缓激肽的羟基之间的反应选择性的影响的图。图7a、7b、7c、7d和7e是显示摩尔过量的加标签试剂对氨基-标记的基团的得率和对过度-标记聚糖的水平的影响的色谱图。这些图证实必须如何优化试剂对葡糖基胺的摩尔过量,以获得高得率(大于80%)而不引入高(大于0.2摩尔%)水平的“过度标记的”物质。图8a、8b和8c是显示通过带有荧光检测的lc分析的自汇集的人igg释放的荧光标记的n-葡糖基胺的结果和猝灭溶液组成对荧光背景水平的影响的色谱图。图8a显示在反应后用加入的乙二胺猝灭后和在用1:14:85甲酸/水/acn洗涤液洗涤后的结果。图8b显示在反应后用加入的异丙胺猝灭后和用1:14:85甲酸/水/acn洗涤液洗涤后的结果。图8c显示在反应后用加入的己胺猝灭后和用1:14:85甲酸/水/acn洗涤液洗涤后的结果。图9a、9b和9c是对标记的葡糖基胺获得的荧光色谱图,显示在spe期间所用的不同洗涤液具有不同的作用。图9a显示用约15:约85体积(15:85(v/v))比例的dmf/acn洗涤后的色谱图。图9b显示用约85%acn洗涤后的色谱图。图9c显示用约1:约14:约85体积(1:14:85(v/v/v))比例的甲酸/水/acn洗涤后的色谱图。图10是使用在此描述的方法,如在实施例2中所述,对用标记试剂-1标记的汇集的人igg葡糖基胺获得的荧光色谱图。图11a是使用在此描述的方法,如在实施例3中所述,对用标记试剂-1标记的西妥昔单抗葡糖基胺获得的荧光色谱图。图11b是使用在此描述的方法,如在实施例3中所述,对应于图11a的基础峰强度色谱图。图12a是从hepes缓冲的反应的spe中产生的对标记试剂-1标记的抗桔霉素鼠igg1葡糖基胺获得的hilic荧光色谱图。图12b是从磷酸盐缓冲的反应的spe中产生的对标记试剂-1标记的抗桔霉素鼠igg1葡糖基胺获得的hilic荧光色谱图。图13a是从标记后反应混合物的直接hilic分析产生的对标记试剂-1标记的抗桔霉素鼠igg1葡糖基胺获得的hilic荧光色谱图。图13b是从标记后反应混合物的在线spe-hilic分析产生的对标记试剂-1标记的抗桔霉素鼠igg1葡糖基胺获得的hilic荧光色谱图。图13c是从标记后反应混合物的高ph/低ph在线spe-hilic分析产生的对标记试剂-1标记的抗桔霉素鼠igg1葡糖基胺获得的hilic荧光色谱图。图14a是rfms标记的来自抗桔霉素鼠igg1的n-聚糖的hilic-flr-ms。图14b是iab标记的来自抗桔霉素鼠igg1的n-聚糖的hilic-flr-ms。图14c显示对rfms和iab标记聚糖的响应因子。图15a是rfms标记的来自汇集的人igg的n-聚糖的hilic-flr-ms。图15b是2ab标记的来自汇集的人igg的n-聚糖的hilic-flr-ms。图15c显示对rfms和2ab标记聚糖的响应因子。图16a和16b显示聚糖标记的相对性能。发明详述本文提供分析生物样品的化合物的新方法。为分析糖基化生物分子,分子经天然或通过合成处理去糖基化,然后在选择的条件下接触标记试剂,以优化的得率和用最少的过度-标记生产衍生的葡糖基胺。无需首先分离样品的生物分子,进行衍生化。色谱分析也可在获得的衍生化混合物上直接进行,这样的分析包括,但不限于,高压液相色谱(“hplc”)、超高效液相色谱(uhplc)、质谱、超临界流体色谱、紫外光(“uv”)和/或荧光(“flr”)检测。然而,分离任选地包括如在此描述的spe离线和spe在线技术。在此描述的方法适合用于自动分析和处理系统例如由hewitsonetal.,在美国专利申请号62/100,252中描述的那些,通过引用结合到本文中。为了广泛种类的应用,包括但不限于蛋白质和肽鉴定和定量、细胞培养基的监测和分析、和食物和饲料的营养含量,本文提供用于高压液相色谱或检测器的这些自动采样和反应系统。自动采样和反应系统可提供齐全可用的分析(turn-keyanalysis),其可被优化用于高压液相色谱处理和检测。公开的方法和系统可以与不同的类型的检测器、tuv、pda或flr检测器一起使用。自动采样和反应系统也可用于应用特定的性能认定,提供相同的结果:不同工作日之间、不同设备之间、全世界的不同实验室之间。此外,在此描述的方法适合用于各种生产和分配自动化的工作流系统和方案。在此描述的方法可以用于液体处理的自动化工作流系统和使用机器人技术的那些,以增加通过量并带给实验室提高的效率和安全性。自动化工作流系统常常用作生物制药、研究和临床诊断的平台并包括,但不限于,数字分配器、dna提取、pcr设置、elisa、多道移液、移液平台和相关的仪器,和被整合以在实验室中产生高效工作流的软件。在本方法中,葡糖基胺在其中蛋白质物质尚未从混合物中耗尽的条件下在溶液中标记。反应混合物的条件包括温度、有机溶剂组成、有机溶剂浓度、缓冲液组成、ph、离子强度、摩尔过量的试剂,且时间被选择和控制,以便可以实现伯胺和葡糖基胺的羟基之间的所需反应选择性。在本文描述的方法中,用于用各种标记试剂(特别是快速加标签的标记试剂)标记葡糖基胺或加标签的条件被优化。如在此所用的,短语“糖基化生物分子”意指并包括蛋白质、肽、聚糖、氨基酸、脂质、dna、rna和核酸。待分析的糖基化生物分子(有时也以单数或以复数称为生物分子或化合物)可天然具有聚糖或可用伯胺、仲胺或叔胺生产聚糖。伯胺和仲胺可以单数或以复数存在。对于在此描述的标记试剂,伯胺和仲胺被认为是“可修饰的”胺。更特别地,胺是具有官能团的有机化合物,所述官能团含有带有孤对电子的氮原子。一般地,胺是氨的衍生物,其中一个或多个氢原子已被取代基例如烷基或芳基替代。示例性的胺包括氨基酸、生物胺、三甲胺和苯胺。氨的无机衍生物在此也被称为胺。此外,糖基化生物分子可在样品内具有单一类型或具有多种类型的混合物。糖基化生物分子可包括,但不限于,胺(伯胺、仲胺等)、氨基酸、肽、蛋白质、聚胺、糖基化化合物、具有聚糖基团的化合物或糖蛋白等。更一般地,糖基化生物分子可意指糖已天然或者经合成处理被加入的任何分子。因此,糖基化生物分子、糖基化化合物或糖蛋白是已经历共翻译或翻译后修饰并含有一个或多个附接于其的碳水化合物和聚糖基团的化合物或分子。这样的糖基化化合物可以是天然存在的或合成产生的并包括蛋白质、脂质、氨基酸(甚至当作为肽和蛋白质中的残基存在时)、抗体、肽或其它有机分子。n-聚糖(或n-连接的聚糖)可附接于天冬酰胺或精氨酸侧链的酰胺或胺基的氮。在其它方面,o-连接的聚糖可附接于丝氨酸、苏氨酸、酪氨酸、羟基赖氨酸或羟基脯氨酸侧链的羟基氧,或附接于脂质上的氧。在本文提出的方法的上下文中,糖基化化合物可以单数或以复数形式存在于样品中。葡糖基胺或n-糖苷,是一类由胺与至碳水化合物的β-n-糖苷键组成的化合物,形成环状半缩醛胺醚键(α-氨基醚)。换言之,葡糖基胺是具有附接于氨基的糖基基团的化合物。葡糖基胺(glycosylamine)可包括,但不限于,核苷例如腺苷,和具有胺基的糖苷例如n,n-二甲基-β-d-吡喃葡萄糖胺、葡糖胺(glucosylamine)、葡糖基-正丁胺、葡糖基-正己胺、葡糖基-正辛胺、葡糖基-正癸胺、葡糖基-正十二烷基胺、麦芽糖基-正十二烷基胺。而且,d-葡萄糖、d-半乳糖、乳糖、纤维二糖和麦芽糖在1当量碳酸氢铵的存在下,在用氨水溶液处理后,将全部产生相应的葡糖基胺、1-氨基-1-脱氧-d-葡萄糖、1-氨基-1-脱氧-d-半乳糖、1-氨基-1-脱氧乳糖、1-氨基-1-脱氧纤维二糖和1-氨基-1-脱氧麦芽糖。特定的糖基化模式已与健康和疾病的状况相关,并且n-聚糖分析被多种工业,包括制药生物技术生产越来越多地应用。许多基于蛋白质的生物药品是糖基化蛋白质,而蛋白质糖基化中的不受控制的变化具有很大的监管问题。例如,各个聚糖结构的相对量在过程开发期间需要被监测以建立制备的生长和纯化步骤的稳定性。n-聚糖(一种形式的葡糖基胺)的荧光标记对检测氨基酸的n-聚糖在生物样品中的分布特性是有益的,因为它改进了检测的灵敏性和选择性以及聚糖的色谱表现。同样地,广泛种类的酶和化学技术可被用于蛋白质去糖基化和聚糖释放。酶去糖基化技术利用糖苷酶,其可包括,但不限于,n-糖苷酶a(pngasea)、n-糖苷酶f(pngasef)、o-糖苷酶、神经氨酸酶、β1-4半乳糖苷酶和β-n-乙酰氨基葡糖苷酶。例如,如在本文详细描述的,糖蛋白样品可以用酶,肽n-糖苷酶f(pngasef)去糖基化,其从糖蛋白除去n-连接的寡糖,除了在还原端上含有α(1-3)-连接的岩藻糖的化合物之外。然而,n-糖苷酶a(pngasea)可除去全部n-聚糖。其它有用的酶包括内切糖苷酶或糖酰胺酶例如内切糖苷酶和n-聚糖酶。一旦酶去糖基化,n-聚糖作为葡糖基胺从天冬酰胺残基释放。为了从糖蛋白化学释放n-聚糖,糖蛋白可以用无水肼于90℃处理数小时。为从糖蛋白释放o-聚糖,糖蛋白用o-聚糖酶处理或用无水肼化学处理,特别是于60℃持续最多6小时。或者,o-聚糖可通过采用用于还原性碱-催化的β-消除、非-还原性β-消除的碱性硼氢化物,或通过使用其它释放剂例如三氟甲烷-硫酸或各种胺释放。其它化学技术包括,但不限于,肼解作用(hydrazynolysis)和三氟甲烷磺酸(tfms)处理。而且,聚糖分析越来越多地应用于生物学研究和临床分析。特定的糖基化模式已与健康和疾病状态相关。而且,糖基化变化可调节蛋白质的生物学活性,如例如对重组免疫球蛋白的fc部分的糖基化所证实的。因此,分析来自糖蛋白的寡糖的方法往往聚焦于相关的和随后衍生的聚糖的分析。然而,尽管这些方法允许独立于载体糖蛋白而深入分析寡糖结构,但不提供关于聚糖的附着位点的信息。因此,蛋白质糖基化概况的特征鉴定具有很大的重要性,因为它对于各种调节目的和生物制药药物的生产都是需要的。释放的聚糖池具有巨大的复杂性和结构的异质性,这需要有效的分离方法和高度灵敏性检测方法。各个聚糖结构的相对量在过程开发期间需要被监测以建立制备的生长和纯化步骤的稳定性。因此,本文提出制备标记葡糖基胺的方法。先前未描述的技术与标记试剂一起使用以快速生产分析备用的(analysis-ready)聚糖。这些技术包括用包含荧光部分、质子亲和力/荷电标签基团和n-羟基琥珀酰亚胺酯或氨基甲酸酯活性基团的标记试剂标记葡糖基胺,所述标记试剂例如如下面表1中的标记试剂-1、标记试剂-2、标记试剂-3、标记试剂-4所鉴定的。表1。可与在此描述的方法组合使用的其它标记试剂包括在美国专利申请号14/458,760(题目为具有增强ms信号的聚糖和其它生物分子的快速荧光标记(rapidfluorescencetaggingofglyacnsandotherbiomoleculeswithenchancedmssignals),未公布)中鉴定的那些标记试剂。见第2页4行至4页9行;11页4行至25页18行和29页1行至30页10行,通过引用结合到本文中。另外的标记试剂也可在以下专利中发现:美国专利号7,148,069(col.8,l.56-col.9,l.54和col.15,l.22-29),通过引用结合到本文中;美国专利号7,494,815(col.7,l.19-col.11,l.24),通过引用结合到本文中;美国专利号8,124,792(col.2,l.13-col.4,l.5和col.7,l.11-col.17,l.20),通过引用结合到本文中;和美国专利号5,296,599(col.4,l.66-col.5,l.32和col.5,l.66-col.7,l.28),通过引用结合到本文中。而且,所述方法可以是制备其它标记葡糖基胺,包括从备选的标记试剂衍生的那些的基础。如在此描述的,这样的标记试剂可具有可供选择的反应性基团,例如异氰酸酯、异硫氰酸酯或亚胺酸酯/硫代亚胺酸酯(thioimidates),和/或备选的官能团例如荷电标记以提高负离子模式质谱分析。选择和控制标记反应的条件,包括温度、有机溶剂组成、有机溶剂浓度、缓冲液组成、ph、离子强度、摩尔过量的试剂,和时间,以便实现伯胺和羟基之间的所需反应选择性。进而,标记聚糖的得率被优化,而所谓“过度标记的”聚糖(由>1个标记修饰的聚糖/葡糖基胺)的生成被最小化。包含含亲水性胺的化合物(乙二胺)的猝灭溶液也可使用。这种猝灭溶液不仅控制允许葡糖基胺与标记试剂反应的时间,而且还将反应的ph移至升高的ph(>10),这提高标记聚糖在高有机溶剂(即>50%乙腈)中的溶解性,从而有利于基于亲水相互作用色谱(“hilic”)的下游spe程序。从糖蛋白释放的聚糖在其中蛋白质物质尚未从混合物中有意耗尽的条件下在溶液中标记。示例性标记反应紧接下面显示。为开发和优化用nhs氨基甲酸酯试剂和/或nhs酯试剂标记葡糖基胺的方法的条件,使n-琥珀酰亚胺基n-甲基氨基甲酸酯,一种小的nhs氨基甲酸酯试剂(标记试剂-4)与单肽,缓激肽反应。这种肽被用作葡糖基胺的代用品,鉴于葡糖基胺分解为具有醛末端的还原糖,其在各独立的实验中不易被研究。tarentino,a.l.etal.,2-iminothiolane:areagentfortheintroductionofsulphydrylgroupsintooligosaccharidesderivedfromasparagine-linkedglyacns,glycobiology1993,3(3),279-85。缓激肽仅含有一个伯胺(其n-末端)以及一个羟基,因而是一种优化用于葡糖基胺衍生化的标记得率和选择性的有用的工具。缓激肽还含有两个高碱性精氨酸残基,使得当n-末端被标记时,有可能通过lc-ms测定反应产物,而无电离效率方面的重大偏差的问题。通过测量%修饰的缓激肽群体(用单胺修饰)以及在其羟基处修饰(单羟基修饰或两个位点修饰)的缓激肽,发明人发现了其中标记得率和对于超过羟基的胺的反应选择性是最优的反应条件。如图2-5中所示,提供%修饰对比摩尔过量的试剂的许多图示。此外,例举了对于这种类型反应的最佳条件的发现,包括:如图2中所示的温度,如图3中所示的有机溶剂组成,如图3中所示的有机溶剂浓度,如图4中所示的缓冲液组成,如图4中所示的ph,和如图5中所示的离子强度。简言之,当:(1)温度在环境至低于-环境温度;(2)二甲基甲酰胺(dmf)被用作有机溶剂;(3)dmf占不超过20-30%的反应混合物;(4)采用在ph7.9和ph8.2之间的磷酸钠溶液缓冲液,和(5)磷酸盐浓度维持在≤50mm时,以最小水平的过量标记获得高标记得率的衍生的化合物。过量标记少于约1摩尔%,更优选约0.0-约0.5摩尔%,和在一些实施方案中约0.0-约0.2%。此外,缓冲液浓度可以是在约5mm-约1000mm之间,或在一些实施方案中为约5mm-约200mm或约5mm-约100mm或约5mm-50mm。可以获得高产率的标记葡糖基胺,其具有超过可修饰胺的约10-约2000,或约30-约1000,或约40-约500;或约50-约300之间范围的量的摩尔过量的标记试剂。而且,尽管在得率方面不必然有利,有机溶剂已显示可用于一些标记试剂例如标记试剂-1的溶解。发明人还发现20-30%dmf足以提高溶解性,而不严重影响标记反应的得率和选择性(图3)。为此理由,包含20-30%dmf的反应混合物是优选的。如图3中提供的,对于二甲亚砜(dmso)和二甲基甲酰胺(dmf)的有机溶剂组成和浓度作为标记反应的共溶剂被测试。dmso和dmf二者是极性非质子溶剂并都有大的介电常数(>20)和大的偶极矩。其它极性非质子溶剂包括四氢呋喃(thf)和乙腈。具有高极性,这些溶剂溶解荷电物质,包括各种阴离子和亲核试剂例如cn(-)和ho(-)。没有氢键,亲核试剂在溶液中是相对“自由的”且更具反应性的。因此,特别可用于本方法中的溶剂包括非-亲核的、极性非质子溶剂。一般地,非质子溶剂的共同特征包括:(1)可接受氢键的溶剂,(2)溶剂没有酸性氢中心(丙酮和酯不满足这一标准);和(3)溶剂溶解有机盐。极性非质子溶剂是将溶解许多盐,但缺乏酸性氢的溶剂。这种类型的溶剂常常具有中间介电常数和极性。然而,如上所注明的,某些极性非质子溶剂既具有高介电常数,又具有高偶极矩,另一个实例是乙腈(“mecn”)和hmpa(六甲基磷酰胺)。为限定最适反应时间和甚至更特别地,为限定反应在其结束前必须被允许继续进行的时间,执行基于缓激肽测定的时程研究。图6显示在通过加入二乙胺终止反应前,来自标记试剂-4与缓激肽反应不同时间长度的时程实验的结果。如在图6中所示,为获得标记化合物与总伯胺浓度的最适摩尔比例,使用500-倍、400-倍、300-倍、200-倍和100-倍摩尔过量的浓度的标记试剂-1进行标记反应。这个时程显示nhs氨基甲酸酯的反应在概述的条件下,在120秒后有效地完成。为了用葡糖基胺标记实施,因此优选在进行另外的样品处理之前,允许nhs氨基甲酸酯反应继续进行2-10分钟。因此,反应时间的范围可在约10秒-约30分钟之间,或在一些实施方案中,反应时间是约30秒-约10分钟或优选地,反应时间是在约2分钟-约5分钟之间。已采用上述方法来标记从几种单克隆抗体,包括鼠igg和从鼠sp20细胞系表达的嵌合igg(西妥昔单抗)释放的n-聚糖。为制备标记的n-聚糖,用肽n-糖苷酶f(pngasef)使糖蛋白样品去糖基,接着与标记试剂于室温下反应。标记试剂溶解于无水二甲基甲酰胺(dmf)中至127mm的浓度。去糖基化混合物和试剂溶液随后以2.5:1体积比混合,以生产包含大约36mm标记试剂和4.8µm释放的n-聚糖(以葡糖基胺的形式)的反应混合物。在这些条件下,葡糖基胺与源自样品的任何其它胺物质(最重要的是前体糖蛋白的蛋白性胺)一起存在于反应混合物中。糖蛋白倾向于含有许多比n-聚糖位点更加蛋白质性的胺(即赖氨酸残基)。因此,去糖基化混合物中最丰富的胺将是蛋白质性胺。发明人还发现,在速度和再现性方面,有意维持样品中的蛋白质性胺是有利的。因此,较高浓度的标记试剂(即标记试剂-1)可加入到反应混合物中,且进而任何其它未说明的胺和亲核试剂对标记的产率具有不重要的影响。结果,葡糖基胺的标记在样品之间将是更可重现的。此外,标记试剂的量可以更容易对胺修饰的所需特征进行调整。例如,高得率的标记葡糖基胺可以更容易地获得,伴有较少的生产过度-标记的聚糖物质的问题。作为备选,标记反应(有时在此也称为“衍生化”或“衍生化步骤”)可以在从去糖基化混合物耗尽蛋白质物质后进行。igg样品含有大约75种蛋白质性胺。因此,基于igg的、含有约4.8µm葡糖基胺(2个聚糖;每个重链上各一个)的释放的聚糖混合物将含有至少180µm的总伯胺组合物。发明人发现在这些条件下,生物样品的所需修饰可通过实施约18-约180mm的标记试剂浓度获得,该浓度代表超过总伯胺浓度的范围为约100-约1000倍摩尔过量的试剂。为生产最佳标记得率和相对低水平的过度-标记聚糖(≤0.24%),采用至少约200-倍摩尔过量的条件是优选的(图7)。一般地,允许标记反应继续进行最少约5分钟。然而标记反应可以较短的时间(即,数秒)发生。通过加入猝灭溶液,标记反应终止。猝灭溶液含有乙二胺/纯水/乙腈的约5:5:90(v/v/v)混合物。通过加入猝灭溶液,通过加入显著浓度的胺(>100mm)以清除剩余的、未反应的标记试剂来终止标记反应。猝灭溶液也将反应混合物的ph从约6.5-约9的ph移至大于10的ph。移动至碱性ph确保标记的葡糖基胺以及反应副产物(其可包括脲-连接的分子)保持可溶性。通过加入猝灭溶液提供的高ph使标记的质子亲和力/荷电标签基团去质子化,因此在高有机/低极性溶液中提高溶解性。这有利于依赖聚糖的极性的样品制备程序,如当基于hilic的spe被用来从标记反应混合物富集标记的聚糖时的情况。yu,y.etal.,arapidsamplepreparationmethodformassspectrometriccharacterizationofn-linkedglyacns,rapidcommunmassspectrom2005,19(16),2331-6。如果溶液的ph不改变,反应副产物和标记葡糖基胺二者可共同沉淀,特别是当在衍生化步骤期间使用磷酸盐缓冲液时。如在以下实施例ii中所述,猝灭溶液以约9:约1(9:1(v/v))的体积比被加入到标记反应混合物中。在该步骤后,终止标记反应,并使反应产物溶解于与hilicspe相容的溶液中。胺在猝灭溶液中的特性对该程序是重要的。胺应是明显亲水性的。乙二胺因此是猝灭溶液的优选的胺。在其它方面,如通过图8的数据所示的,其它疏水性胺例如异丙胺和己胺已被测试并发现当样品经带有荧光检测的lc分析时,产生由于反应副产物所致的不需要的水平的背景。为分离衍生的葡糖基胺,亲水性spe吸附剂,例如waterssep-pakaminopropyl、watersoasishlb,或watersoasiswax,可以被调节用于spe,此后用来处理猝灭的、acn-稀释的样品。waterssep-pakaminopropyl柱芯具有中等极性的、具有弱碱性表面的基于二氧化硅的键合相。该吸附剂可以被用作具有对酸性/碱性分析物的不同选择性的极性吸附剂,或用作在低于ph8的含水介质中的弱阴离子交换剂。对于这种类型的柱芯的应用包括酚和酚类色素、石油馏分、糖和药物和代谢物的提取。柱芯被设计附接于用于可靠的、正压流的泵和注射器以加快样品处理时间。柱芯还配有可拆卸的贮库并在真空辅助流设备和某些自动系统上使用。spe可同样地用小型化的96-孔格式化装置执行。装载后,spe吸附剂可以经洗涤,以进一步促使反应副产物的移除。促进从反应副产物选择性地富集标记葡糖基胺的条件已被发现。发明人发现涉及1:14:85(v/v/v)甲酸/水/乙腈的spe洗涤步骤有效减少反应副产物,当检测器饱和无效峰和倾斜基线时,其在获得的样品的lc色谱图中得到证明。可用于这个方面的其它hilicspe洗涤溶剂包括0.3:14.7:85(v/v/v)磷酸/水/乙腈。有用的hilicspe洗涤溶剂可在酸组成中变化,例如,当有机溶剂组成是约50-约100%体积时,从约0.01至约10%变化,或当有机溶剂组成是约60-约98%体积时,从约0.05至约5%,或当有机溶剂组成是约70-约96%时,从约0.3至约3%变化,当有机溶剂组成是约80-约95%体积时,从约0.1至约3%变化。在spe期间已采用几种洗涤条件。如在图9a、9b和9c中所示,洗涤条件对标记葡糖基胺获得的荧光色谱图具有影响。1:14:85(v/v/v)甲酸/水/乙腈有效减少荧光背景并且还有效将荷载样品的ph从高ph移至低ph,鉴于据报道高ph引起n-乙酰基葡糖胺残留物差向异构化为n-乙酰基甘露糖胺,这是一个重要的考虑因素。liu,y.etal.,investigationofsamplepreparationartifactsformedduringtheenzymaticreleaseofn-linkedglyacnspriortoanalysisbycapillaryelectrophoresis,analchem.2009,81(16),6823-9。用酸性1:14:85(v/v/v)甲酸/水/乙腈洗涤溶液洗涤快速移动荷载样品的ph也确保基于二氧化硅的spe吸附剂的溶解最小化。pettersson,s.w.etal.,chemicalstabilityofreversedphasehighperformanceliquidchromatographysilicaundersodiumhydroxideregenerationconditions,jchromatogra2007,1142(1),93-7。如在此描述的,高ph/低phhilicspe处理减少在标记葡糖基胺的分析期间遭遇的色谱背景。然而,色谱背景和hilic柱的无效时间附近的洗脱峰已被发现通过使用hepes(2-[4-(2-羟基乙基)哌嗪-1-基]乙磺酸)而非磷酸盐来缓冲标记反应,进一步减少强度。这已导致具有较少强度的色谱背景的spe洗脱液。见图12a和12b。hepes有利地改变了分布、溶解性,或试剂副产物的spe选择性并可以这样做而无需猝灭溶液。因此,hepes缓冲的溶液可以用于去糖基化和释放的n-葡糖基胺的随后的标记。此外,可以使用缓冲两性离子化合物(像hepes),并具有约7-约9之间的pka的为非-亲核的其它缓冲液,包括,但不限于ada(n-(2-乙酰胺基)-2-亚氨基二乙酸)、bes(n,n-双(2-羟基乙基)-2-氨基乙磺酸)、bicine(n,n-双(2-羟基乙基)甘氨酸)、dipso(3-(n,n-双[2-羟基乙基]氨基)-2-羟基丙烷磺酸)、epps(4-(2-羟基乙基)-1-哌嗪丙烷磺酸)、hepbs(n-(2-羟基乙基)哌嗪-n′-(4-丁烷磺酸))、mobs(4-(n-吗啉代)丁烷磺酸)、mops(3-(n-吗啉代)丙烷磺酸)、mopso(3-(n-吗啉基)-2-羟基丙烷磺酸)、pipes(1,4-哌嗪二乙磺酸)、popso(哌嗪-n,n′-双(2-羟基丙烷磺酸))。缓冲化合物为中性或阳性(而非阴性)的电离状态可进一步减少色谱背景。因此,可使用阳离子的、非-亲核缓冲化合物,例如叔胺:tea(三乙基氨)、bis-tris(2,2-双(羟基甲基)-2,2′,2″-次氮基三乙醇)、bis-tris丙烷(1,3-双[三(羟基甲基)甲基氨基]丙烷)。此外,在线spe可备选地用于替代离线spe。在线spe通常以一维捕集-洗脱色谱的形式执行,其中所谓“捕集”柱经由流体切换机制与分析柱配对。捕集柱通常包含大颗粒吸附剂,以使高线性速度加载可以实现,同时在线spe的流出物转向废物。样品被注射到捕集/在线spe柱上并洗掉基质成分和污染物。随后,来自捕集柱的流出物被导向分析柱并展开梯度,以洗脱和分离样品的成分。为帮助实现最佳的色谱,捕集柱的静止相必须表现出比分析柱的静止相更低的保持力。捕集和分析柱之间的这种匹配确保分析物再聚集于分析柱上。从而梯度洗脱使分离具有与通过直接注射分析物至分析柱上另外获得的分辨力相匹配的分辨力。通常实施捕集-洗脱色谱用于反相分离,但不用于hilic分离。同样,发明人已发现hilic静止相保持力用于一种新的类型的分析物:聚糖,包括标记聚糖、葡糖基胺,或标记葡糖基胺。用于最佳在线spe/捕集-洗脱色谱的hilic吸附剂和静止相也已被发现。特别是,发明人已发现聚合物的、亲水亲脂平衡(hlb)吸附剂(watersoasishlb)显示出对于捕集静止相的理想的聚糖保持力。特别地,发明人发现在大约10:90水/乙腈的条件下,oasishlb保留聚糖。同时,聚糖开始从具有11-15%水的含水强度的oasishlb洗脱。这种hilic保持力是独特的,因为当与通常用于聚糖hilic色谱的静止相,例如酰胺-键合或多元醇(多羟基)键合静止相的保持力比较时,要弱大约10%乙腈。oasishlb是不仅包含非极性部分,而且还包含重复亲水性酰胺部分的聚合物吸附剂。这种结构为它提供了显著的hilic保持力,其已被发现对捕集/在线spe柱是有利的。更特别地,对于葡糖基胺(聚糖)的在线spe,oasishlb捕集柱(即2.1x30mmwatersoasishlb,20μm粒径)可以与用酰胺键合的有机二氧化硅颗粒(即2.1x50mm,watersglycanbehamide130å1.7μm)填充的分析柱配对。聚糖被荷载到在线spe/捕集柱上,同时流出物通过一些流体学机制被转移到废物中。标记葡糖基胺吸收到spe柱中的吸附剂并以用高有机流动相,例如88%acn以相对快速的流速洗涤。流动相组合物也可用添加剂例如12:88(v/v)的1%的强氨溶液/h2o:acn改变。来自oasishlbspe柱的流出物在洗涤步骤后可以被重新导向分析hilic柱。在洗涤步骤期间,衍生化副产物和样品基质的其它成分被排出到废物中。因此,洗涤经设计以防止干扰。此外,通过使用在线spe,洗涤溶液通常与吸附剂接触只有很短的时间,进一步避免了基质干扰。在线spe还减少了分析师必须花费执行手工样品制备技术的时间量。然后,色谱条件例如在实施例2中描述的和适合聚糖的梯度洗脱的那些条件可被用来获得分离和色谱图。实施例1用于最大化标记得率和最小化过度-标记物质的条件的开发标记试剂(即标记试剂1-4)与10µm缓激肽反应。反应在不同的条件下进行;所有反应通过加入5m二乙胺hcl溶液(ph9.8)猝灭,以使最终的二乙胺浓度为大约800mm。获得的反应产物通过lc-uv-ms,使用与waterssynaptg2-s组合的watersacquityuplch-classbio测定。将样品以10µl的体积注入到acquityuplccshc18c18,130å,1.7µm,2.1x50mm柱中。于60℃的温度下并以0.3ml/min的流速,使用包含在水中的0.1%tfa(v/v),在acn中的0.1%tfa(v/v),在acn中的0.1%fa(v/v),和在acn中的0.1%fa(v/v)的流动相之间的四元梯度执行分离。洗脱物质通过uv吸光度(10hz,214nm)和通过电喷雾离子化质谱(100-2000m/z,2hz)检测。通过以上处理获得的数据在图2-6中呈现。watersacquityuplch-classbio是设计有非-不锈钢材料的生物-惰性流路径,以保持大的生物分子完整并通过色谱柱移动的色谱系统。它利用专门打算用于分析蛋白质、肽、核酸和聚糖的低于2-µm混合粒子化学。它具有流量通过针头注射器(flow-through-needleinjector)和四元溶剂传递系统,提供运行多个色谱模式(包括,但不限于,反相(rp)、离子交换(ex)、尺寸排阻(sec)或亲水性相互作用(hilic)色谱)的能力。这种色谱系统使用允许二元、三元或四元梯度操作与任选加入的用于待使用的额外溶剂的六端口溶剂选择阀的多-溶剂共混。此外,这种系统允许用户改变流动相调节剂和有机溶剂的浓度。代替手工制备溶剂混合物,这种系统使得纯溶剂和浓缩储备液按需要共混。当ph和离子强度由用户指定时,它也提供自动调节和计算对所需条件需要的缓冲储备液的比例。而且,系统减少体积并使频带展宽最小化以维持高效率的分离。它使用超低分散检测器(ultra-low-dispersiondetectors)以维持能够用于生物学应用的多个检测需求的峰的完整性。此外,系统使用各种各样的加热器和多-柱管理器(柱切换),以提供用于控制色谱选择性和最高保留时间精度的低分散和精确的温度管理。watersacquityuplch-classbio可与waterssynaptg2-si高清晰度质谱组合。waterssynaptg2-si是使用离子的碰撞截面(ccs)特性的高清晰度质谱系统,通过使用t-波离子迁移以提高生物分子分析的峰容量、特异性和灵敏性。waterssynaptg2-si装配有较大的离子采样孔口、增强真空泵配置和离子传输光学器件。这种二元-t-波、离轴设计将来自离子源的离子高效率转移到四极ms分析仪,同时确保不需要的中性污染物被过滤掉。这增加了ms离子强度,同时使背景噪音最小化。与其它反相uplc柱比较,watersacquityuplccshc18(苯基-己基和氟代-苯基)柱提供替代的选择性。这些柱使用15%碳负载,c18化学,具有2.1mm内径、30mm长度、封端的,提供具有混合颗粒基质的球形颗粒形状,例如混合有机/无机颗粒。它们含有130å孔径和低硅醇活性并能够在从约ph1-约ph11的范围内处理ph。实施例2从汇集的人igg和鼠igg1释放的标记葡糖基胺的快速制备以类似的方式制备汇集的人igg(从血清)和鼠igg1的样品。igg的冻干样品在50mm磷酸钠(ph7.9)中重新构成并与肽n-糖苷酶f(pngasef,newenglandbiolabs,p0705)混合。制备这种混合物以使igg浓度是大约1mg/ml和pngasef的活性浓度是大约25单位/µl。随后,将这种混合物于37℃孵育1小时(产生鼠igg的fc区的完全去糖基化的条件)。在1小时孵育完成后,使去糖基化混合物冷却至室温,此后用等体积的50mm磷酸钠(ph7.9)稀释。同时,使标记试剂-1溶解于无水二甲基甲酰胺(dmf)中至127mm的浓度(假定标记试剂-1被纯化为与nhs的1:1复合物)。去糖基化混合物和试剂溶液随后以2.5:1体积比混合,以生产包含大约36mm标记试剂-1和4.8µm释放的n-聚糖(和180µm的总伯胺浓度)和反应副产物(包括,但不限于,对应于标记试剂的胺水解产物和由这种胺和标记试剂产生的反应产物(即,脲连接的分子)的化合物)的反应混合物。反应被允许进行5分钟,然后通过加入猝灭溶液(5:5:95(v/v/v)乙二胺/水/acn)终止。猝灭溶液以9:1的体积比加入到反应混合物中。此后,生成的猝灭的、acn-稀释的样品经历spe,使用waterssep-pakaminopropylµelution板和真空驱动的spe。首先用水清洁各孔,然后用15:85(v/v)水/acn调节。随后,将猝灭的acn-稀释的样品加载到各孔。吸附的样品然后用包含1:14:85(v/v/v)甲酸/水/acn的溶液洗涤。最后,富集的、标记葡糖基胺使用包含100mm乙酸铵(ph7)、5%acn的洗脱剂从spe吸附剂洗脱。获得的标记的葡糖基胺以spe洗脱液的形式直接分析,或者相反作为干燥(通过离心蒸发)并重新构成的样品分析。标记葡糖基胺的分析经由hilic分离和荧光和质谱检测的组合(watersacquityuplch-classbiosystem与waterssynaptg2-s质谱配对)进行。用1.7µm酰胺-键合有机二氧化硅颗粒填充的2.1x50mm柱与包含50mm甲酸铵(ph4.5)的含水流动相(流动相a)和包含acn的流动相(流动相b)共同使用。含水样品作为1µl体积注射并于60℃的温度,根据梯度表2分离。标记的葡糖基胺使用荧光检测器(5hz,激发λ=265nm,发射λ=425nm)和通过电喷雾离子化质谱(600-2500m/z,1hz)检测。图10呈现了代表从这种程序得到的样品的色谱数据。表2时间(min)流速(ml/min)%a%b%c%d曲线初始0.4257500初始150.4524800615.50.2100000616.50.2100000617.70.2257500619.20.4257500621.70.42575006实施例3从西妥昔单抗释放的标记葡糖基胺的快速制备采用在实施例2中所述类似的程序,除了采用独特的去糖基化处理,从西妥昔单抗,一种从鼠sp20细胞表达的嵌合igg1制备标记葡糖基胺。图11a和图11b提供了代表从这种程序产生的西妥昔单抗样品的色谱数据。实施例4图13a、13b和13c提供用标记试剂-1标记的n-葡糖基胺(通过带有watersoasishlb的在线spe纯化,然后使用用酰胺-键合静止相填充的分析柱分离)获得的实施例hilic荧光色谱图。可以使用对watersoasishlb的备选方案。由二氧化硅或有机二氧化硅基质颗粒构成的未键合或二醇-键合的吸附剂已被发现显示出将使得它们为有用的捕集静止相选择的保持力。更特别地,图13b是对通过具有50mm甲酸铵(ph4.5)的流动相a和acn的流动相b的在线spe纯化的n-葡糖基胺获得的色谱图。而且,紧接下面的表3是其spe梯度表。图13c是对通过具有50mm甲酸铵(于ph4.5)的流动相a,acn的流动相b和1%(v/v)氢氧化氨的流动相c的在线spe纯化的n-葡糖基胺获得的色谱图。表4是其spe梯度表。表3表4如在此描述的,可以使用亲水亲脂平衡聚合物吸附剂的变体,包括,但不限于phenomenexstratax(一种具有用于分析物-吸附剂相互作用的多个保留模式的含有n-乙烯基吡咯烷酮的官能化聚合物吸附剂)、thermohyperseppep(用脲官能团修饰的多孔dvb材料填充的柱芯)、agilentsampliqopt(用酰胺修饰的二乙烯基苯聚合物树脂填充)、watersoasiswax、watersoasiswcx、watersoasismax,和watersoasismcx。phenomenexstratax吸附剂是针对中性和芳族分析物的具有表面积800sq.m/g的基于聚合物的反相官能化吸附剂,提供在侵袭性的高有机洗涤条件下中性、酸性或碱性化合物的保留。这种吸附剂依赖于3种保留机制:π-π键合、氢键合(偶极-偶极相互作用)和疏水性相互作用。这种类型的吸附剂是耐ph的并可处理0-14的ph范围。thermohypersepretainpep柱芯用脲官能团修饰的多孔聚苯乙烯dvb材料填充,所述材料可提供极性和非-极性分析物的回收。这些柱提供具有30-50µm粒径和30mg床重量的吸附剂。watersoasis吸附剂是spe吸附剂的家族,目前以商品名oasis销售。这些吸附剂在ph极端和在宽范围的溶剂中是稳定的。这些吸附剂具有良好的极性化合物的保留性,和相对疏水性保留能力为传统的基于二氧化硅的spe吸附剂像c18的3倍。另外的提取产品包括产品例如watersoasishlb,一种用于酸性、中性和碱性化合物的通用吸附剂。oasishlb是从特定比例的两个单体,亲水性n-乙烯基吡咯烷酮和亲脂性二乙烯基苯制得的亲水-亲脂-平衡的、水-可湿润的、反相吸附剂。watersoasiswax柱芯是已被优化用于强酸性化合物的高选择性的聚合物反相、弱阴离子交换、水-可湿润的混合-模式聚合物吸附剂。watersoasiswcx(弱阳离子交换)是由共聚物基质制得的混合-模式、水-可湿润的spe吸附剂,其提供全部类型的分析物,尤其是强碱(pka>10)和季胺的保留性。保留机制是混合模式(离子交换和反相二者)。watersoasismax(混合-模式阴离子交换)是被优化获得用于提取具有阴离子交换基团的酸性化合物(pka<1)的较高选择性和灵敏性的混合-模式聚合物吸附剂。这种吸附剂是水-可湿润的。watersoasismcx(混合-模式阳离子交换)是优化用于保留具有pka2-10的碱性化合物的混合-模式聚合物吸附剂。实施例5标记n-聚糖的hilic-荧光-esi-ms(ms/ms)分析为评价响应因子,标记的n-聚糖经由hilic分离结合荧光和质谱检测,使用uhplc色谱仪(acquityuplch-classbio,waters,milford,ma)分析。使用用1.7µm酰胺-键合有机二氧化硅颗粒(acquityuplcglycanbehamide130å,waters,milford,ma)填充的2.1x50mm或者2.1x150mm柱以及包含50mm甲酸铵(ph4.4)的含水流动相和包含acn的另一种流动相。样品作为1µl含水体积或10µlacn/dmf体积注射并于60℃根据梯度分离。标记n-聚糖使用荧光检测器(5hz扫描率,gain=1,acquityuplcflr,waters,milford,ma),使用激发和发射波长检测。洗脱聚糖也通过阳离子模式电喷雾离子化质谱,使用离子迁移率能力qtof质谱仪检测(synaptg2-s,waters,milford,ma),用3.0kv的毛细管电压,120℃的源温度,350℃的去溶剂化温度,和80v的样品锥电压操作。以1hz的频率,大约20,000的分辨率,获得在500-2500m/z的范围内的质谱。下表5提出与在这个实施例中描述的聚糖标记相关的结构和缩写,以及所用的试剂。表5。结果和讨论高灵敏性荧光和ms检测已评价rfms标记提供n-聚糖分析的灵敏性。特别是,rfms标记聚糖的响应因子已成为针对用备选的试剂标记的聚糖的响应因子的基准。最密切相关的、可市售获得的对rfms的替代选择是氨基苯甲酰胺或iab的nhs氨基甲酸酯类似物(cook,k.s.etal.,biologicals,40(2),109-17(2012)。图14a和14b呈现对于等量的从鼠igg1单克隆抗体释放的并用rfms和iab分别标记的n-聚糖的hilic荧光和基础峰强度(bpi)ms色谱图。基于观察到的色谱峰面积,对于在igg概况中的最丰富的聚糖,岩藻糖基化双分枝的fa2聚糖(oxford注释),确定了荧光和ms检测的响应因子(图14c)。harvey,d.etal,proteomics,9(15),3796-801(2009);glycobase3.2http://glycobase.nibrt.ie(2015年1月6日访问)。图14a是rfms的hilic-flr-ms的结果和图14b是来自抗桔霉素鼠igg1的iab标记的n-聚糖的结果。荧光(flr)色谱图和基础峰强度(bpi)ms色谱图被显示。使用用1.7μm酰胺键合有机二氧化硅(130å)静止相填充的2.1x50mm柱分离标记聚糖(自0.4µg糖蛋白,1µl含水注射液)。如在图14c中所示,对于rfms和iab标记聚糖的响应因子(作为每份从1µg抗桔霉素鼠igg1产生的n-聚糖样品的fa2峰面积测量)。荧光(flr)和ms(基础峰强度)响应因子被分别显示。分析按一式两份进行。发明人的对于fa2聚糖的结果表示rfms标记的聚糖比用iab标记的n-聚糖生产2倍的更高的荧光信号,且更令人吃惊地,接近800倍的更大的ms信号。以类似的方式,rfms标记也与常规的2-ab标记比较。为描绘这样的比较,从具有rfms或者2-ab的汇集的人igg制备的n-聚糖通过以等效质量负载的hilic-flr-ms分析(分别为图15a和15b)。图15a描述rfms的hilic-flr-ms和图15b描述来自汇集的人igg的2-ab标记n-聚糖。荧光(flr)色谱图和基础峰强度(bpi)ms色谱图被显示。使用用1.7μm酰胺-键合有机二氧化硅(130å)静止相装填的2.1x50mm柱,分离标记的聚糖(~14pmol总聚糖、1µl含水注射液)。fa2聚糖的量经由两点外部校准(two-pointexternalcalibrations),用定量标准(rfms衍生的丙胺和2-ab标记的三乙酰基壳三糖)校准。在图15c中,用于rfms和2-ab标记的聚糖的响应因子(作为通过外部校准测定的每皮摩尔的fa2的fa2峰面积测量)。荧光(flr)和ms(基础峰强度)响应因子被分别显示。分析按一式两份进行。鉴于快速标记和还原性胺化通过明显不同的程序进行,外部校准使用定量标准建立,以测定从hilic柱荷载和洗脱的fa2聚糖的量。使用这些校准量的fa2聚糖计算的响应因子在图15c中提供。与2-ab标记聚糖对比,rfms标记聚糖以优越的灵敏性,特别是以14倍更高的荧光和160倍更大的ms信号被检测。为总结以上的观察结果,发明人已绘制了iab和2-ab的响应因子作为针对rfms的响应因子的百分比(图16)。图16显示聚糖标记性能的相对百分比(%)。响应因子显示为对比rfms标记n-聚糖的荧光和ms响应因子的百分比。比较结果从公布的n-聚糖的比较外推,其中已发现当与2-ab比较时,普鲁卡因酰胺提供可比较的荧光和多达50倍的更大的esi-ms灵敏性。荧光和ms灵敏性的获得在该图中是明显的,因为它描绘了归一于rfms响应因子的iab和2-ab响应因子。在图16中,也提供用另一个备选的标记试剂,普鲁卡因酰胺的还原性胺化的相对性能。普鲁卡因酰胺是氨基苯甲酰胺的化学类似物,其最近已显示当它们通过hilic-esi(+)-ms分析时,提高还原性胺化的聚糖的离子化。先前的研究已显示当与2-ab标记聚糖比较时,普鲁卡因酰胺标记的聚糖获得可比较的荧光信号,和10-50倍更大的ms信号,该观察由发明人自身的2-ab和普鲁卡因酰胺标记的n-聚糖分析证实。klapoetke,s.etal.,j.pharmbiomedanal53(3),315-24(2010)。与普鲁卡因酰胺比较,rfms因此被预测提供,最小3-倍的ms灵敏性的增加。由于这两种标记含有叔胺部分,提示rfms标记聚糖的优越的电离作用源自比普鲁卡因酰胺标记更疏水的rfms标记是合理的。确实,先前的研究已显示对聚糖标记添加疏水性表面积导致增加的电喷雾电离。walker,s.h.etal,jamsocmassspectrom22(8),1309-17(2011);bereman,m.s.etal.,chemcommun(camb)26(2),237-9(2010)。除了相对疏水性核心结构外,rfms标记还具有强碱性侧链,因此是值得注意的。更值得注意的是,以上响应因子数据提示rfms标记提供对于n-聚糖的hilic色谱特性的前所未有的荧光和ms灵敏性。实施例6氨基酸的快速标记的预示方法在此描述的方法和试剂可被用来执行氨基酸的快速标记。与适宜的缓冲剂和稀释剂一起,可以进行氨基酸的柱前衍生化(pre-columnderivatization)和分析。为重新构成粉末形式的衍生试剂,在设置于55℃的加热块或其它装置上加热试剂和稀释剂的小瓶。重新构成的试剂通常在乙腈中在10mm的范围内。重新构成的试剂可于室温下贮存通常至多1周。然而,可以利用备选的溶剂和温度。为衍生氨基酸样品,将60µl的硼酸盐缓冲液加入到在6x50mm样品管中重新构成的样品中并涡流。然后加入20µl的重新构成的试剂并立即涡流数秒钟。允许孵育混合物,通常于室温下1-5分钟。然后将试管中的内容物转移至小瓶并用含有硅树脂衬隔膜的盖密封。将小瓶于55℃加热10分钟。如果紧密封闭并防止蒸发,氨基酸衍生物可于室温下贮存至多1周。因此,虽然设计了葡糖基胺的制备,所述方法可应用于氨基酸标记(不管氨基酸残基是游离的或是蛋白质和肽中的成分)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1