烟草及烟草制品中农药残留量的测定方法与流程
本发明涉及一种烟草及烟草制品中农药残留量的测定方法,属于农药残留检测领域。
背景技术:
目前,烟草生长过程中施用的农药主要包括杀菌剂和杀虫剂。杀菌剂常用的有多菌灵、腈菌唑、戊菌灵、霜霉威、甲基硫菌灵、三唑醇、三唑酮和嘧菌酯,杀虫剂常用的有啶虫脒和吡虫啉。鉴于上述农药在烟叶中的高残留风险,严格控制烟草及烟草制品中农药的残留量是保证烟草制品吸食安全性的关键。国际烟草科学研究中心(CORESTA)于2003年提出对烟草中99种农药给予指导性残留限量,并于2013年将监控指标扩充到120种。
口含烟制品是新型烟草制品的重要形式之一,不会产生“环境烟草烟气”。国外口含型无烟气烟草制品的常用添加剂包括甜味剂、酸碱调节剂、香精香料等,其制作工艺的多样性造成该类制品在外观、物理表征尤其是化学指标方面的较大差异。袋装口含型无烟气烟草制品是将磨碎的烟草颗粒加工后直接装入小袋中,使用时将其放于口腔中,无需咀嚼,烟草成分经唾液溶解后通过口腔粘膜吸收。由于农药残留普遍存在于烟草中,而烟草是口含烟的主要原料,因此口含烟中不免有农药残留。但是口含烟的种类较多,基质差别各异,无法采用空白基质配标的方法进行定量分析。
同位素内标稀释法能够降低基质效应对定量结果的影响(同位素稀释质谱法测定奶粉基体中药物残留的基质效应研究,杨总,2013;同位素稀释-固相萃取-LC/MS/MS法测定婴幼儿配方食品中双酚类化合物,高梦婕等,分析化学),同时LC-MS/MS法具有较高的检测灵敏度以及较强的专属性,无需使分析物之间完全色谱分离,样品前处理操作简单,分析测试时间短,可实现多类型多残留农药的同时分析。公布号CN105699538A的发明专利公开了一种卷烟主流烟气中常用农药含量的测定方法,包括:1)样品前处理:在烟气捕集气相物中加入乙腈和内标,漩涡振荡提取后再依次加入无水硫酸镁、氯化钠、柠檬酸钠和柠檬酸二氢钠,漩涡振荡提取后离心取上清,在上清中加入无水硫酸镁和PSA吸附剂(N-丙基乙二胺),漩涡振荡提取后离心取上清,过滤,稀释定容后待测;2)超高效液相色谱-串联质谱测定:将待测溶液注入UPLC-MS/MS系统,色谱条件:UPLC HSST3色谱柱,100×2.1mm,粒径1.8μm,流动相A:0.1%甲酸水溶液,流动相B:0.1%甲酸甲醇溶液,流速:0.4mL/min,柱温:35℃,进样量:2μL,洗脱条件:0~2min(90%A-50%A),2~2.4min(50%A-30%A),2.4~4min(30%A-20%A),4~6min(20%A-5%A),6~9.8min(5%A-5%A),9.8~10min(5%A-90%A),10~12min(90%A-90%A);质谱条件:电喷雾离子源,喷雾电压:2.6kV,离子化温度:350℃,雾化气流量:800L/Hr,锥孔气流量:50L/Hr,碰撞气流量:0.15mL/min,碰撞气为氩气,其余气体为氮气,驻留时间:30msec,采集模式:正离子MRM,监测离子对及其对应的碰撞能量见表1。该专利采用乙腈浸提,并利用基质分散固相萃取法提取和净化样品,经UPLC-MS/MS分析测定得到烟气中仲丁灵、多菌灵、吡虫啉、稻瘟灵、甲霜灵、腈菌唑、二甲戊灵、霜霉威、甲基硫菌灵、三唑酮和三唑醇的残留量,该方法检测灵敏度高,选择性强,但是对烟草中色素等成分的去除效果欠佳,且仅采用TPP一种内标,不能去除基质效应的干扰不能进行准确定量。
技术实现要素:
本发明的目的是提供一种烟草及烟草制品中农药残留量的测定方法。
为了实现以上目的,本发明所采用的技术方案是:
烟草及烟草制品中农药残留量的测定方法,包括样品的前处理步骤,具体为:在样品中加入同位素混合内标溶液,平衡后加入乙腈进行提取,再加入无水硫酸镁、氯化钠、柠檬酸钠和柠檬酸氢二钠纯化,纯化后离心取上清液,在上清液中加入C18和中性氧化铝净化,净化后过滤,即得待测溶液。
所述烟草制品如口含烟制品等。
所述同位素混合内标溶液的配制可采用以下步骤:分别将各农药相应内标的标准品(如氘代物)与乙腈混合后定容,得到各农药相应内标的标准储备液;分别取一定量的各农药相应内标的标准储备液,混合后用乙腈定容,得到同位素混合内标溶液。
所述提取、纯化、净化均可采用漩涡振荡的方式,如在转速1800~2200rpm下漩涡振荡1~6min。
所述提取中每1~2g样品加入5~10mL乙腈。
所述纯化中每1~2g样品加入3~5g无水硫酸镁、1g氯化钠、1g柠檬酸钠和0.5g柠檬酸氢二钠。
所述净化中每1mL上清液加入40~60mg C18和40~60mg中性氧化铝。C18和中性氧化铝均可购自安捷伦科技。
所述农药包括多菌灵、腈菌唑、戊菌唑、霜霉威、甲基硫菌灵、三唑醇、三唑酮、啶虫脒、吡虫啉、嘧菌酯等。
烟草及烟草制品中农药残留量的测定方法,还包括农药残留量的测定步骤,具体为:将待测溶液注入液相色谱-串联质谱系统,以内标法定量分析,经计算得到烟草制品中农药的残留量;液相色谱条件为:色谱柱:Poroshell 120EC-C18,规格:100mm×3.0mm,粒径2.7μm;流动相A:0.1%甲酸乙腈溶液(体积分数),流动相B:10mM甲酸铵水溶液,流速:0.4mL/min,柱温:40℃,进样量:10μL,洗脱条件(具体见下表1):0~5min,90%A-60%A,5~10min,60%A-40%A,10~15min,40%A-10%A,15~20min,10%A-10%A,20~20.1min,10%A-90%A,20.1~30min,90%A-90%A;串联质谱条件:电喷雾离子源,离子化温度:500℃,电离电压:5500V,扫描方式:正离子扫描,MRM模式采集(参数见下表2),雾化气流量:60psi,气帘气流量:20psi,辅助加热气流量:60psi,碰撞气流量:12psi,四种气体均为氮气。
表1流动相梯度洗脱条件
表2各农药及其同位素内标监测离子对、去簇电压及碰撞能量
注:嘧菌酯与吡虫啉性质相似,二者均以吡虫啉-d4为内标;其他农药均以各自对应的氘代物为内标。
所述内标法定量分析为:将系列浓度的混合标准工作溶液注入液相色谱-串联质谱系统,以各农药标准品与其相应内标的色谱峰面积的比值对应其浓度作回归分析,得到标准曲线;将待测溶液注入液相色谱-串联质谱系统,得到各检出农药与其相应内标的色谱峰面积的比值,代入标准曲线,即得待测溶液中各农药的浓度。
所述系列浓度的混合标准工作溶液的配制可采用以下步骤:分别将各农药的标准品与乙腈混合后定容,得到各农药的标准储备液;分别取一定量的各农药的标准储备液,混合后用乙腈定容,得到混合标准储备液;用乙腈将上述混合标准储备液稀释成具有浓度梯度的混合标准工作溶液,稀释过程中分别加入同位素混合内标溶液,即为系列浓度混合标准工作溶液。
本发明的有益效果:
本发明中样品前处理操作简单、快捷,杂质及干扰物的去除效率高,能大大缩短前处理时间。样品经乙腈浸提后先用无水硫酸镁、氯化钠、柠檬酸钠和柠檬酸氢二钠提纯,再用C18和中性氧化铝净化,能够有效去除烟草中色素及烟草植物组织的其他共萃物(如脂肪酸、叶绿素等),避免样品浓缩、净化、衍生等操作造成待测农药组分的损失。
本发明中烟草制品农药残留量的测定方法具有以下优点:1)由于口含烟种类繁多,基质差别各异,无法采用空白基质配标的方法进行定量分析,而同位素内标稀释法能够降低基质效应对定量结果的影响;2)操作简单、快捷,检测灵敏度高,重复性好,检测限在0.002~0.011μg/g之间,加标回收率为85%~98%,平均相对标准偏差小于6%。
附图说明
图1为本发明中农药残留量测定方法的流程示意图;
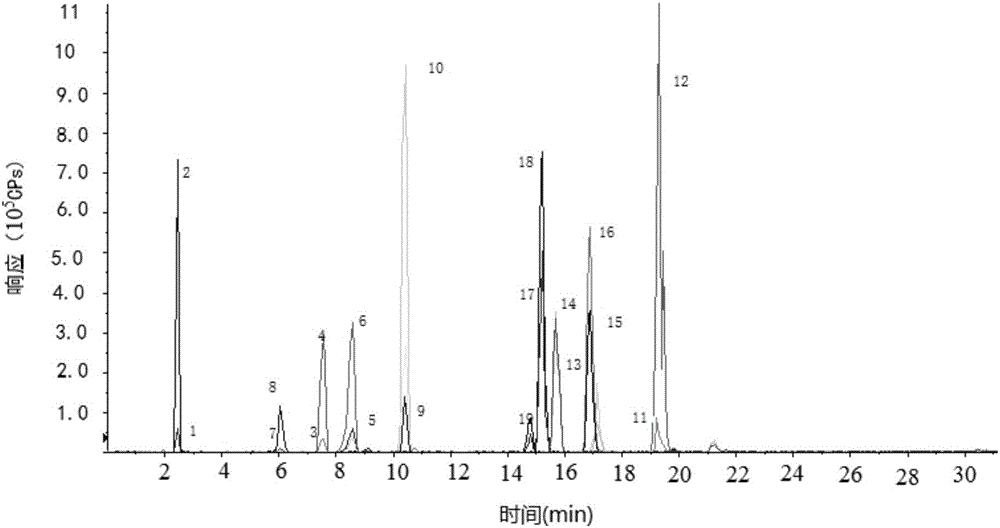
图2为实施例1中混合标准工作溶液4的离子色谱图;
图3为实施例1中待测溶液的离子色谱图。
具体实施方式
下述实施例仅对本发明作进一步详细说明,但不构成对本发明的任何限制。
仪器与试剂:API 4000四极杆串联质谱仪(美国AB公司);涡旋振荡仪(美国Labnet公司);Sigma 3-30K离心机(德国Sigma公司);AE 163电子天平(感量0.0001g)与AE 166电子天平(感量0.01g)(瑞士Mettler公司)。农药均为标准品;乙腈、丙酮、甲酸均为农残级。
实施例1
口含烟中农药残留量的测定方法(流程示意图见图1),包括以下步骤:
1)样品的前处理
样品的提纯:准确称取1.0g口含烟样品于50mL具塞离心管中,捣碎,加入100μL10μg/mL同位素混合内标溶液(溶液中各内标浓度均为10μg/mL),并置于阴凉干燥处平衡24小时,以与被测物质均匀混合且交换完全,平衡后加入10mL乙腈,然后将离心管置于漩涡混合振荡仪上,以2000rpm速率振荡5min,再向离心管中加入3g无水硫酸镁、1g氯化钠、1g柠檬酸钠和0.5g柠檬酸氢二钠(柠檬酸氢二钠的离子数较柠檬酸二氢钠多,盐析效果更好),立即置于漩涡混合振荡仪上,以2000rpm速率振荡5min,然后以6000rpm速率离心3min。
样品的净化:移取1mL离心后的上清液于1.5mL离心管中,并加入50mg C18和50mg中性氧化铝,于漩涡混合振荡仪上以2000rpm速率振荡2min,再以6000rpm速率离心3min,吸取上清液,经0.45μm有机相滤膜过滤,得到待测溶液。
同位素混合内标溶液的配制步骤为:分别称取10mg(精确至0.1mg)各农药相应内标的标准品于10mL容量瓶中,用乙腈溶解并定容至刻度,得到各农药相应内标的标准储备液;分别移取一定量的各农药相应内标的标准储备液于同一个100mL的容量瓶中,用乙腈定容至刻度,得到10μg/mL的同位素混合内标溶液,于避光、-18℃下保存即可。
2)农药残留量的测定
系列浓度的混合标准工作溶液的配制步骤为:分别称取10mg(精确至0.1mg)各农药的标准品于10mL容量瓶中,用乙腈溶解并定容至刻度,得到各农药的标准储备液;分别移取一定量的各农药的标准储备液于同一个100mL的容量瓶中,用乙腈定容至刻度,得到10μg/mL的混合标准储备液;分别移取混合标准储备液20μL、50μL、100μL、250μL、500μL、1000μL于6个10mL的容量瓶中,并分别加入100μL上述同位素混合内标溶液,用乙腈定容,得到具有浓度梯度的混合标准工作溶液。
混合标准工作溶液中各农药标准品的浓度见下表3。
表3系列浓度的混合标准工作溶液中各农药标准品的浓度
将系列浓度的混合标准工作溶液分别注入液相色谱-串联质谱系统,液相色谱条件为:色谱柱:Poroshell 120EC-C18,规格:100mm×3.0mm,粒径2.7μm;流动相A:0.1%甲酸乙腈溶液(体积分数),流动相B:10mM甲酸铵水溶液,流速:0.4mL/min,柱温:40℃,进样量:10μL,洗脱条件(见表1);串联质谱条件:电喷雾离子源,离子化温度:500℃,电离电压:5500V,扫描方式:正离子扫描,MRM模式采集(参数见表2),雾化气流量:60psi,气帘气流量:20psi,辅助加热气流量:60psi,碰撞气流量:12psi,四种气体均为氮气;测得各农药标准品峰面积和其相应内标峰面积的比值与其浓度关系的一元线性回归方程(各农药的保留时间及相关参数见下表4),混合标准工作溶液4的选择离子色谱图见图2。
将待测溶液注入液相色谱-串联质谱系统,液相色谱及串联质谱条件同上,得到各检出农药峰面积和其相应内标峰面积的比值,代入标准曲线,得到待测溶液中各农药的浓度,其中嘧菌酯30ng/mL,多菌灵28ng/mL,选择离子色谱图见图3),再经计算得到口含烟中各农药的残留量,结果为嘧菌酯0.3μg/g,多菌灵0.28μg/g。
表4各农药的保留时间及相关参数
实施例2
烟叶中农药残留量的测定方法,包括以下步骤:
1)样品的前处理
样品的提纯:将干烟叶粉碎,准确称取2.0g样品于50mL具塞离心管中,捣碎,加入100μL 10μg/mL同位素混合内标溶液(同实施例1),并置于阴凉干燥处平衡24小时,平衡后加入10mL乙腈,然后将离心管置于漩涡混合振荡仪上,以2000rpm速率振荡5min,再向离心管中加入5g无水硫酸镁、1g氯化钠、1g柠檬酸钠和0.5g柠檬酸氢二钠,立即置于漩涡混合振荡仪上,以2000rpm速率振荡5min,然后以6000rpm速率离心3min。
样品的净化:移取1mL离心后的上清液于1.5mL离心管中,并加入50mg C18和50mg中性氧化铝,于漩涡混合振荡仪上以2000rpm速率振荡2min,再以6000rpm速率离心3min,吸取上清液,经0.45μm有机相滤膜过滤,得到待测溶液。
2)农药残留量的测定
将待测溶液注入液相色谱-串联质谱系统,液相色谱及串联质谱条件同实施例1,得到各检出农药峰面积和其相应内标峰面积的比值,代入实施例1的标准曲线中,得到待测溶液中各农药的浓度,其中啶虫脒120ng/mL,多菌灵165ng/mL,再经计算得到烟叶中各农药的残留量,结果为啶虫脒1.2μg/g,多菌灵1.65μg/g。
在本发明的其他实施例中,提取、纯化及净化用试剂的加量均可适当调整,提取、纯化及净化操作中各参数也可适当调整。
试验例
加标回收测试:为判断实施例1中测定方法的准确性,特在样品中加入浓度0.10μg/g的混合标准溶液(与同位素混合内标溶液一起加入),同上进行样品前处理及后续测定操作,加标回收测试结果见下表5。
精密度测试:在样品中加入浓度0.10μg/g的混合标准溶液,同上进行样品前处理及后续测定操作,共进行5次重复试验,结果见下表5。
表5回收率和精密度测试结果
- 还没有人留言评论。精彩留言会获得点赞!