一种人工合成多环麝香的在线分析方法与流程
本发明属于分析化学领域,具体涉及一种人工合成多环麝香的在线分析方法。
背景技术:
人工合成多环麝香以其优雅的芳香气味,优良的定香能力以及低廉的价格,广泛应用于化妆品、洗涤用品等产品中。人工合成多环麝香具有较强的亲脂憎水性,在环境中难降解,易生物富集。开展环境中人工合成多环麝香的高效分析,对了解人工麝香在环境中的分布、传输、迁移以及降解具有至关重要的意义。目前,人工合成多环麝香主要采用c18萃取柱或萃取盘离线富集净化,结合气相色谱-质谱联用技术进行分析。开发功能化材料,籍此建立人工合成多环麝香的在线高效便捷的检测方法备受关注。
整体柱,又称连续床,是近年来新兴的一类微流色谱分离柱。hayes等研究提出了硅胶基聚合介质,即机械性能和耐溶剂性能良好的硅胶整体床层。硅胶整体聚合固定相骨架具有良好的刚性,具有μm级通孔刚性骨架结构和nm级分离中孔,网络渗透性,机械强度高,耐热性和耐溶剂性好,在反相和亲水作用色谱中应用广泛,引起了人们的极大关注。
有机-无机杂化硅胶是一种新兴的聚合功能材料。其中最具代表性的是多面体纳米低聚倍半硅氧烷(poss),该物质具有多个(n=8~12等)立体分布的交联聚合位点,极易于引入各种活性有机功能单体或功能基团。通过聚倍半硅氧烷端基的不饱和基团的聚合反应,高度交联形成具有3d结构的立体化聚合功能材料,所形成的硅胶基整体聚合固定相中含有由si-o交替连接的笼型硅氧骨架无机内核,通过反应性基团与聚合物之间可以发生接枝或者聚合反应,功能基团均匀的分布在poss纳米颗粒表面,拥有高度交联的立体结构,作用位点多、比表面积大、作用效率高。
目前,在疏水性硅胶基固定相的制备方面,文献报道主要通过丙烯酸酯类单体和有机硅氧烷前驱体低交联度交联共聚,芳香性单体聚合固定相研究主要集中在丙烯酸酯类有机聚合整体柱研究领域。引入笼型聚倍半硅氧烷,通过纳米尺寸粒子均匀性地分散于基体材料中,共聚卤代芳香性单体功能单体和卤代芳香性单体二次深度交联反应,开发含有纳米尺寸材料的高疏水性杂化硅胶整体柱,籍此建立人工麝香残留的高效富集、分离方法,具有良好的应用前景。
技术实现要素:
本发明的目的在于提供一种人工合成多环麝香的在线分析方法,该方法采用电色谱高压电场和液相泵高压双重驱动作用,实现人工合成多环麝香快速在线富集、洗脱,并以反相液相色谱实现分离,快速简便,能实现对疏水性人工合成多环麝香的高效分析。
为实现上述目的,本发明采用如下技术方案:
一种人工合成多环麝香的在线分析方法,其主要采用微流电色谱和微流液相色谱组成的二维色谱体系进行分析,即先在第一维的微流电色谱中,以高疏水型杂化硅胶整体柱为载体,在柱同步施加高压电场和液相泵高压,利用此双重驱动作用实现人工合成多环麝香的在线富集,然后将第一维微流电色谱洗脱出的样品切换进入第二维的微流液相色谱,以丙烯酸酯聚合整体柱进行人工合成多环麝香的液相色谱分离;
其具体分析条件包括:以体积比为10:90的乙腈-水为溶液配制样品,样品进样体积为300μl;第一维微流电色谱中以体积比90:10的乙腈-乙酸铵缓冲溶液为流动相,分离电压为+15kv,泵压为1000psi,流速为0.02ml/min;第二维微流液相色谱中以体积比为90:10的甲醇-水为流动相,泵压为1000psi,流速为0.10ml/min;
所述乙酸铵缓冲溶液的ph值为7.00,其中乙酸铵的浓度为5.0mmo1/l。
所述丙烯酸酯聚合整体柱为十八烷基甲基丙烯酸酯聚合整体柱。
所述高疏水型杂化硅胶整体柱由具有3d交联结构的聚倍半硅氧烷与卤代芳烃单体、离子型单体经共价聚合后,再以路易斯酸为催化剂引发卤代芳烃相互之间发生亲核取代反应,形成以聚倍半硅氧烷笼型无机内核为骨架、芳环深度交联结构为作用点的高疏水型改性硅胶基整体柱;
其中,所述卤代芳烃单体为4-乙烯基苄氯;
其中,所述离子型单体为3-磺酸丙基甲基丙烯酸钾盐;
所述聚倍半硅氧烷为聚八甲基丙烯酸酯基笼型倍半硅氧烷;
所述路易斯酸为三氯化铁。
所述高疏水型杂化硅胶整体柱的制备方法包括以下步骤:
(1)采用0.1mol/l的hcl溶液冲洗毛细管空柱30min,再用去离子水冲洗15min,然后用0.1mol/l的naoh溶液冲洗2h,再用去离子水洗至中性,最后用甲醇冲洗15min,氮气吹干;
(2)将甲醇与甲基丙烯酰氧基丙基三甲氧基硅烷按体积比1:1混合,加入步骤(1)预处理好的毛细管中,60℃反应12h,然后甲醇冲洗15min,70℃下氮气吹干;
(3)将卤代芳烃单体、离子型单体、聚倍半硅氧烷与致孔剂按比例混合,超声振荡15min后氮气吹扫10min,然后将该混合液注入步骤(2)的毛细管中,60℃水浴反应12h,再以甲醇为流动相冲洗平衡10~15h;
(4)将惰性卤代烷烃试剂通入步骤(3)制备好的整体聚合固定相中,室温静置2h,然后通入路易斯酸溶液,将毛细管两端封口,置于80℃恒温水浴锅中反应24h,然后依次以甲醇、水、甲醇为流动相,在液相色谱泵上冲洗2h,除去残留反应液,即得。
按质量百分数之和为100%计,步骤(3)所述混合液中各组分的质量百分数为:卤代芳烃单体22.8%~25.8%,离子型单体0.2%,聚倍半硅氧烷4.0%~7.0%,致孔剂70%;
所述致孔剂为十二醇和甲苯的混合物,其中甲苯占混合物质量的27.5%~45.0%。
步骤(4)中所述惰性卤代烷烃试剂为1,2-二氯乙烷;所述路易斯酸溶液为0.1g三氯化铁在1,2-二氯乙烷中的饱和溶液。
本发明的显著优点在于:
(1)本发明提出的一种用于人工合成多环麝香在线分析的新方法,其是采用微流电色谱和微流液相色谱组成的二维色谱体系进行分析,即先在第一维的微流电色谱中,利用电色谱中的高压电场驱动作用(高压电场驱动力形成的区带为平面梯形),辅助一定的液相泵高压,使色谱填料中的流体线速度较大,协同保持了人工合成多环麝香在高疏水型杂化硅胶整体柱上的高效富集。与现有色谱柱上富集模式比较,其流体区带更加平整,压力降变化幅度减小,以有效减少纯液相技术中憎水性人工合成多环麝香在高疏水性富集柱上迁移速率较低而产生的浓度扩散、区带信号展宽等不利影响,实现人工合成多环麝香在高疏水型杂化硅胶整体柱上的高效富集;联用第二维的微流液相色谱,以丙烯酸酯聚合整体柱为载体,分离介质疏水性适中、稳定性高,二者联用有利于实现人工合成多环麝香的快速分离和灵敏检测。
(2)本发明所述的高疏水型杂化硅胶整体柱,是以聚倍半硅氧烷与卤代芳烃单体形成的聚合物,每个笼型聚倍半硅氧烷单体分子上具有八个支化独立分布的烯基作用位点,均可立体地固定化卤代芳烃,具有高疏水性、耐溶剂性特点;并以路易斯酸催化引发固定化的卤代芳烃相互之间产生亲核取代反应,以形成笼型硅氧骨架和交替连接的苯环结构共存的高度交联聚合结构,提高了疏水基团修饰和富集效率。
附图说明
图1为所得高疏水型杂化硅胶整体柱的性能测试情况图。
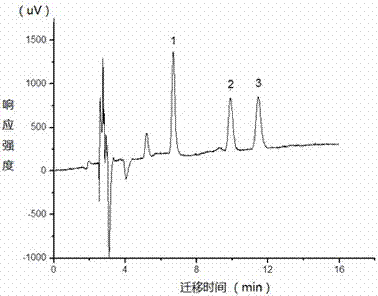
图2为含不同种人工合成多环麝香样品经富集分离后的微流液相色谱图。
具体实施方式
为了使本发明所述的内容更加便于理解,下面结合具体实施方式对本发明所述的技术方案做进一步的说明,但是本发明不仅限于此。
实施例
(1)采用0.1mol/l的hcl溶液冲洗毛细管空柱30min,再用去离子水冲洗15min,然后用0.1mol/l的naoh溶液冲洗2h,再用去离子水洗至中性,最后用甲醇冲洗15min,氮气吹干;
(2)将甲醇与甲基丙烯酰氧基丙基三甲氧基硅烷按体积比1:1混合,加入步骤(1)预处理好的毛细管中,60℃反应12h,然后甲醇冲洗15min,70℃下氮气吹干;
(3)按表1a-d所述比例分别将卤代芳烃单体4-乙烯基苄氯、离子型单体3-磺酸丙基甲基丙烯酸钾盐、聚倍半硅氧烷聚八甲基丙烯酸酯基笼型倍半硅氧烷与致孔剂(由十二醇与甲苯组成)混合,超声振荡15min后氮气吹扫10min,然后将该混合液注入步骤(2)的毛细管中,60℃水浴反应12h,待反应完成后以甲醇为流动相冲洗平衡10~15h,除去毛细管内残留反应试剂;
(4)将1,2-二氯乙烷通入步骤(3)制备好的整体聚合固定相中,室温静置2h;以0.1g三氯化铁在1,2-二氯乙烷中的饱和溶液为催化剂,将其通入上述聚合固定相中,将毛细管两端封口,置于80℃恒温水浴锅中反应24h,然后依次以甲醇、水、甲醇为流动相,在液相色谱泵上冲洗2h,除去残留反应液,即得高疏水型杂化硅胶整体柱。
表1高疏水型杂化硅胶整体柱固定相中聚合组分配比
应用实施例1
使用实施例中制备的整体柱d,以不同体积配比的乙腈-乙酸铵盐缓冲液(乙酸铵浓度为5.0mmol/l,ph7.0)为流动相,分离电压+15kv,泵压力1000psi,流速0.1ml/min,选用非极性的甲苯、丁苯、戊苯、极性硫脲四种中性小分子作为分析对象(保留情况:戊苯>丁苯>甲苯>极性硫脲),测定所得整体柱的疏水性。
图1为所得高疏水型杂化硅胶整体柱的性能测试情况图,其中,1为硫脲,2为甲苯,3为丁苯,4为戊苯。由图1可见,当乙腈含量提高到95%时,相对保留物的出峰顺序仍未发生改变,说明所述整体柱表面的疏水性强。
应用实施例2
使用实施例中制备的整体柱d,以体积比为10:90的乙腈-水为溶液配制样品,样品进样体积300μl;第一维的微流电色谱中以体积比90:10的乙腈-乙酸铵缓冲溶液(乙酸铵浓度为5.0mmo1/l,ph值为7.00)为流动相,分离电压+15kv,泵压1000psi,流速0.02ml/min;第二维的微流液相色谱方法以体积比为90:10的甲醇-水为流动相,泵压力1000psi,流速0.02ml/min,对含不同种人工合成多环麝香的样品进行富集分离。
图2为含不同种人工合成多环麝香样品经富集分离后的微流液相色谱图,其中,洗脱峰1为开司米酮,洗脱峰2为萨利麝香,洗脱峰3为吐纳麝香。由图2可见,不同种多环麝香均可在线得到良好分离。
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
- 还没有人留言评论。精彩留言会获得点赞!