一种注射剂中甘油含量的检测方法与流程
本发明涉及一种药物含量检测方法,尤其是一种注射剂中甘油含量的检测方法。
背景技术:
甘油是日常美容保湿用品,也是注射剂常用辅料,主要用作增溶剂和渗透压调节剂。作为美容用品,主要涂抹在皮肤表面,如面部,手足,身体四肢,并无任何副作用。注射剂主要有静脉注射、肌内注射、皮下注射、皮内注射和脊椎腔注射等给药途径。研究表明,注射液中的高浓度甘油经以上途径进入人体,会出现不同程度的不良反应及副作用。主要原因在于,高浓度甘油具有脱水性,对中枢神经和渗透屏障有直接作用,可以增加血容量,以致引起溶血、头晕、恶心等症状。这些症状在妊娠、高血压、糖尿病、肾病等血容量或血压本身就比较高的情况下,就更加明显。美国食品药品监督管理局(foodanddrugadministration,fda)规定了不同给药途径注射剂中甘油的最大使用限量,皮下注射剂为32.5%,静脉滴注剂为22.5%,肌肉注射剂的最大用量为15.36%,皮内注射剂为1.6%,静脉注射剂为2.5%。
甘油为注射剂常用辅料,目前报道的甘油的测定方法有高碘酸氧化滴定法、gc-fid、hplc-rid和hplc-elsd法等。高碘酸钠滴定法虽然简单快捷,但其专属性和准确度较差。若待测样品含有邻羟基类成分、能被高碘酸钠氧化的成分、能与氢氧化钠溶液反应的成分,均会影响测量结果的准确性。采用气相(gc)检测法测定甘油时,甘油的沸点高(290℃),不易沸出。hplc-rid法示差折光检测器准确度低,数据处理十分繁琐。elsd检测器的稳定性较差,且操作要求高,一般在避光,室内恒温,恒速气流下检测。
定量质子核磁共振(quantitativeprotonnuclearmagneticresonance,q1h-nmr)技术是一种日趋成熟的仪器分析方法,现已在化学、药物、食品、农业以及军事领域有广泛的应用,且我国2010版药典将q1h-nmr收载在附录中。q1h-nmr用于药物分析的最大优势在于可同时提供化合物结构确证的定性信息和含量测定的定量信息。与hplc方法相比,该方法具有样品预处理步骤简单、测试速度快、不破坏样品等优点,能准确地检测出注射剂中的甘油成分,为注射剂中甘油成分的含量测定提供了一种新的手段。
注射剂在制备工艺上往往需要添加一些辅料,例如助溶剂、抗氧剂、ph调节剂、等渗调节剂、膨松剂等。近年来注射剂安全问题层出不穷,辅料的安全问题日益受到关注。鉴于此,对注射剂中的甘油成分进行定性定量分析,对保证注射剂安全有效具有重要意义。
技术实现要素:
本发明所要解决的技术问题在于提供一种注射剂中甘油含量的检测方法。
为解决上述技术问题,本发明的技术方案是:
一种注射剂中甘油含量的检测方法,基于定量核磁共振技术(1h-nmr和13c-nmr)对注射液中的甘油成分进行含量测定,具体步骤为:
1h-nmr:
(1)内标溶液的配制
称取马来酸标准品与内标物tsp于容量瓶中,用d2o(重水)溶解并定容至刻度线,用于所有实验溶剂;
(2)自制供试品溶液的制备
分别精密称取乳酸钠标准品、甘油,加入超纯水、内标溶液,得自制供试品溶液,将所述供试品溶液可转移至5mm核磁管中,直接用于1h-nmr测试;
(3)注射液样品溶液的制备
分别精密移取注射液0.1ml,加入2ml内标溶液,转移至5mm核磁管中,加封直接用于1h-nmr测试;
(4)1h-nmr指纹图谱的测定
将核磁管置于核磁共振仪中,采用水峰抑制脉冲序列测定注射液样品溶液的1h-nmr指纹图谱和自制品溶液的1h-nmr图谱,其中测试参数及条件为:抑水峰序列zgcppr,观测频率600.25mhz,测定温度298k,谱宽16384.0hz,90°脉冲,采样数据点65536,采样时间1.7039s,扫描次数16次,采样前弛豫延迟时间16s,接受增益28;
(5)样品1h-nmr图谱分析
选择δ3.657-3.608作为定量质子峰的积分区间;选择内标马来酸δ6.511-6.293为定量质子峰的积分区间;tsp其信号峰在δ0.00,用作定零点,排除内标物对待测成分定量峰的影响;
(6)1h-nmr甘油的含量测定
在上述实验条件下采集图谱,对甘油和内标物的定量峰分别进行积分,按照1h-nmr内标法根据积分结果依照下式计算样品的质量:
其中,px为待测物的浓度(mmol/l),pma为内标马来酸的浓度(mmol/l),ax为待测物中定量峰面积,ama为内标马来酸的定量峰面积,nx为待测物的定量质子数,nma为内标马来酸定量质子数;
13c-nmr:
(1)内标溶液的配制
称取马来酸标准品于容量瓶中,用d2o(重水)溶解并定容至刻度线,用于所有实验溶剂;
(2)自制供试品溶液的制备
分别精密称取乳酸钠标准品、甘油,加入超纯水、内标溶液,得自制供试品溶液,将所述供试品溶液可转移至5mm核磁管中,直接用于13c-nmr测试;
(3)注射剂供试品溶液的制备
精密移取注射液并加入上述内标溶液中溶解,将上述供试品溶液可转移至5mm核磁管中,直接用于13c-nmr测试;
(4)13c-nmr指纹图谱的测定
将核磁管置于核磁共振仪中,采用门控去耦序列测定注射液样品溶液的13c-nmr指纹图谱和自制品溶液的13c-nmr图谱,其中测试参数及条件为:门控去耦序列zgig,观测频率603.80mhz,测定温度298k,谱宽36057.7hz,90°脉冲宽度16.28μs,采样数据点16384,采样时间0.4544s,扫描次数32次,采样前弛豫延迟时间10s,接受增益2050;
(5)样品13c-nmr图谱分析
根据13c-nmr谱,选择内标马来酸δ135.62处的官能团上的碳原子为定量峰,甘油δ73.07处的碳原子作为定量峰,分别对峰面积和峰强度进行积分和记录;
(6)13c-nmr甘油的含量测定
在上述实验条件下采集图谱,对甘油和内标物的定量峰分别进行面积和峰强度积分,按照13c-nmr外标法根据积分结果依照下式计算样品的质量,
y=ax+b;
其中x代表定量成分定量峰的积分面积(或峰强度)与内标定量峰的积分面积(或峰强度)之比,y代表定量成分与内标的摩尔浓度比,a和b分别为标准曲线方程的系数。
优选的,上述注射剂中甘油成分的含量检测方法,所述1h-nmr步骤(1)中所述内标溶液中含有22.67mmol/l马来酸、0.59mmol/ltsp(例如,精密称取马来酸标准品131.54mg,置于10ml棕色容量瓶中,用d2o溶解(含tsp0.59mmol/l)并定容至刻度,获得马来酸浓度为22.67mmol/l的内标溶液);所述13c-nmr步骤(1)中所述内标溶液中含有543.11mmol/l马来酸(例如,精密称取马来酸标准品630.40mg,置于10ml棕色容量瓶中,用d2o溶解并定容至刻度,获得马来酸浓度为543.11mmol/l的内标溶液)。
优选的,上述注射剂中甘油成分的含量检测方法,所述1h-nmr步骤(2)中自制供试品溶液,方法如下:精密称取每1.44mg乳酸钠标准品,1.25mg甘油,加入30μl超纯水,600μl内标溶液,作为自制供试品溶液;所述13c-nmr步骤(2)中自制供试品溶液,方法如下:精密称取每14.64mg乳酸钠标准品,12.05mg甘油,加入480μl超纯水,120μl内标溶液,作为自制供试品溶液。
优选的,上述注射剂中甘油成分的含量检测方法,所述1h-nmr步骤(5)中甘油和马来酸的信号进行归属如下表1;所述13c-nmr步骤(5)中甘油和马来酸的信号进行归属如下表2。
表1
表2
优选的,上述注射剂中甘油成分的含量检测方法,所述待检测的注射液为消痔灵注射液、甘油果糖氯化钠注射液、参芎葡萄糖注射液或依托咪酯乳状注射液。
优选的,上述注射剂中甘油成分的含量检测方法,所述注射剂供试品溶液的制备方法如下:精密移取注射液并加入所述内标溶液中溶解,其中,
每0.1ml消痔灵注射液,用0.3ml超纯水稀释,加入0.1ml内标溶液,得消痔灵供试品溶液;
每0.1ml甘油果糖氯化钠注射液,用0.3ml超纯水稀释,加入0.1ml内标溶液,0.3ml超纯水,得甘油果糖氯化钠供试品溶液;
每0.4ml参芎葡萄糖注射液,加入0.1ml内标溶液,得参芎葡萄糖供试品溶液;或
每0.4ml依托咪酯乳状注射液,加入0.1ml内标溶液,得依托咪酯乳状供试品溶液。
上述注射剂中甘油成分的含量检测方法,所述1h-nmr检测过程中选择δ3.657-3.608作为定量质子峰的积分区间,是因为选择与样品中其它质子峰分离较好的特征峰作为待测成分的定量峰时,对于甘油定量应考虑其在样品中与其他成分的重叠效应,甘油果糖氯化钠注射液中,甘油在δ3.80-3.75处的峰与样品中其他成分有重叠;参芎葡萄糖注射液中δ3.80-3.75及δ3.57-3.52处的峰与其他成分存在重叠;因此,为保证积分的一致性与准确性,对所述注射液的甘油统一选择δ3.657-3.608作为定量质子峰的积分区间;
样品1h-nmr图谱分析过程中,根据质子信号峰的化学位移、偶合裂分情况,及相关标准品的核磁共振1hnmr谱,鉴定甘油有3组质子信号,包括δ3.77(1h,m)2位上连氧次甲基质子信号和δ3.63(2h,dd,j=11.7,6.5hz)1、3位上两个连氧亚甲基质子信号及δ3.54(2h,dd,j=11.7,4.4hz)为1、3位上两个连氧亚甲基质子信号;马来酸有1组质子信号,δ6.40(2h,s)为2、3位双键质子信号;将得到的1hnmr图谱应用mestrenova9.0软件定标,仔细校正基线与相位,选择δ3.657-3.608作为定量质子峰的积分区间;选择内标马来酸δ6.511-6.293为定量质子峰的积分区间;tsp其信号峰在δ0.00,用作定零点,排除内标物对待测成分定量峰的影响;
所述13c-nmr检测过程中鉴定甘油中共2组碳原子信号,包括δ73.1,2位碳原子信号和δ62.5,1、3位碳原子信号;马来酸共2组碳原子信号,包括δ132.6,2、3位双键碳原子信号和δ170.8,1、4位羰基碳原子信号;门控去耦是全去藕图谱,可以为碳原子定量提供信息,但不是待测组分中所有官能团上的碳原子都可以作为定量计算的选择。由于本次实验d1时间的限制,甘油与马来酸两种分子中不同碳原子t1的不同及门控去耦存在的部分noe效应,导致响应效果的差异。甘油中2组碳原子信号的峰强度(或面积)与其个数成正比,而马来酸δ171.77的碳原子与δ135.62的碳原子,由于其所处的环境不同,与氢核相近的碳原子的信号被增强,故马来酸δ135.62的峰强度(或峰面积)与碳原子数成正比,δ171.77的峰强度(或峰面积)与碳原子数不成比例。因此,选择内标马来酸δ135.62处的官能团上的碳原子为定量峰,甘油δ73.07处的碳原子作为定量峰,分别对峰面积和峰强度进行积分和记录。
本发明的有益效果是:
上述注射剂中甘油含量的检测方法,首次使用定量质子核磁共振技术用于检测注射剂中甘油的含量,并且采用1h-nmr和13c-nmr两种定量角度,可以有效解决注射液中甘油成分的定量分析问题,所述方法具有样品预处理步骤简单、测试速度快、稳定性好、精密度高、重现性好、准确度高、专属性强、不破坏样品等优点,能准确地检测出注射剂中的甘油成分,为注射剂中甘油成分的含量测定提供了一种新的手段,在实现注射剂中甘油成分质量检测方面具有广阔的应用前景。
附图说明
图1为自制样品溶液和4种注射液样品溶液的甘油质子峰放大1h-nmr图谱,其中,a-自制样品溶液、b-甘油果糖氯化钠注射液、c-参芎葡萄糖注射液、d-消痔灵注射液、e-依托咪酯乳状注射液;
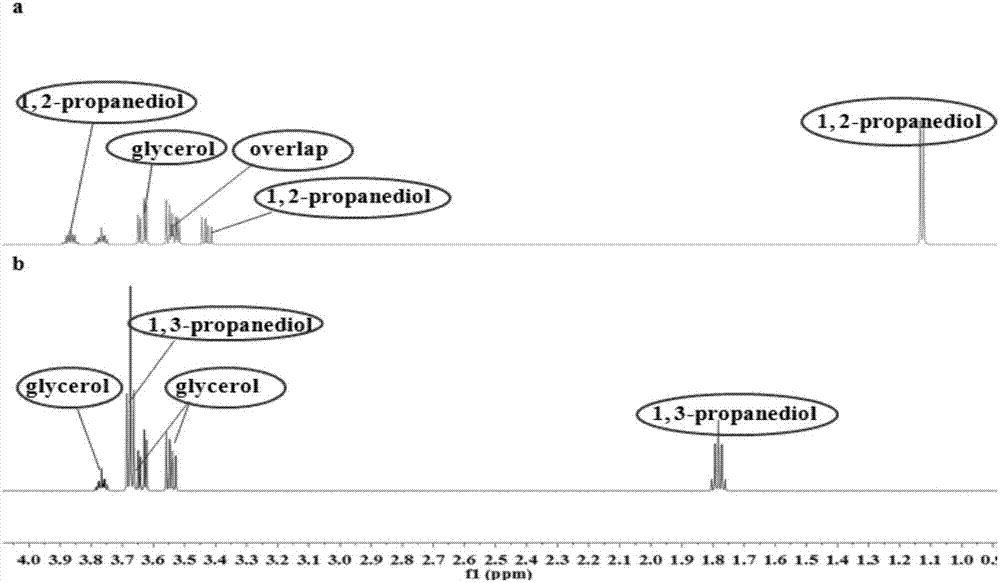
图2为专属性样品1h-nmr图谱,其中,a-甘油与1,2-丙二醇、b-甘油与1,3-丙二醇;
图3为专属性样品13c-nmr图谱,其中,a-甘油与1,2-丙二醇、b-甘油与1,3-丙二醇。
具体实施方式
为了使本领域的技术人员更好的理解本发明的技术方案,下面结合具体实施方式对本发明所述技术方案作进一步的详细说明。
实施例1
四种注射液1h-nmr标准指纹图谱的建立
1.1仪器与试药
brukeravanceⅲ600型超导核磁共振波谱仪,质子激发频率600.25mhz,配置bbfo正相观察宽带探头和bvt3200数字控温仪;5mm标准核磁样品管,美国norell公司生产;mettlertoledoxp6天平(瑞士mettler公司)。
d2o氘代度≥99.9%,美国sigma-aldrich公司;3-(三甲基甲硅烷)丙酸-d4钠盐(tsp)氘代度>98%,sigma-aldrich公司生产;甘油(glycerol,c3h8o3),≥99.5%,美国sigma-aldrich公司生产;马来酸(maleicacid,c4h4o4),≥99.94%,美国sigma-aldrich公司生产;乳酸钠(sodiumlactate,c3h5nao3),≥99.0%,美国sigma-aldrich公司生产;1,2-丙二醇(1,2-propanediol,c3h8o2),≥99.5%,美国sigma-aldrich公司生产;1,3-丙二醇(1,3-propanediol,c3h8o2),≥98%,美国sigma-aldrich公司生产。
四种注射液名称、生产厂家及生产批号分别为:甘油果糖注射液,购自南京正大天晴制药有限公司,生产批号1504242;参芎葡萄糖注射液,购自贵州景峰注射剂有限公司,生产批号20150682;消痔灵注射液,购自吉林省吉安市益盛药业股份有限公司,生产批号15050505;依托咪酯乳状注射液,购自江苏恩华药业股份有限公司,生产批号20150803。
1.2自制供试品溶液的制备
称取马来酸标准品与内标物tsp于容量瓶中,用d2o(重水)溶解并定容至刻度线制得内标溶液,所述内标溶液中含有22.67mmol/l马来酸、0.59mmol/ltsp。
精密称取1.44mg乳酸钠标准品,1.25mg甘油,加入30μl超纯水,600μl内标溶液,得自制供试品溶液,将所述供试品溶液可转移至5mm核磁管中,直接用于1h-nmr测试。
1.3四种注射液供试样品溶液的制备
分别精密移取四种注射液(甘油果糖注射液,参芎葡萄糖注射液,消痔灵注射液,依托咪酯乳状注射液)各0.1ml,加入2ml内标溶液,转移至5mm核磁管中,加封直接用于1h-nmr测试。
1.41h-nmr指纹图谱的测定
将核磁管置于核磁共振仪中,采用水峰抑制脉冲序列测定注射液样品溶液的1h-nmr指纹图谱和自制品溶液的1h-nmr图谱,其中测试参数及条件为:抑水峰序列zgcppr,观测频率600.25mhz,测定温度298k,谱宽16384.0hz,90°脉冲,采样数据点65536,采样时间1.7039s,扫描次数16次,采样前弛豫延迟时间16s,接受增益28。
1.5样品1h-nmr图谱分析
如图1所示,根据质子信号峰的化学位移、偶合裂分情况,及相关标准品的核磁共振1hnmr谱,鉴定甘油有3组质子信号,包括δ3.77(1h,m)2位上连氧次甲基质子信号和δ3.63(2h,dd,j=11.7,6.5hz)1、3位上两个连氧亚甲基质子信号及δ3.54(2h,dd,j=11.7,4.4hz)为1、3位上两个连氧亚甲基质子信号;马来酸有1组质子信号,δ6.40(2h,s)为2、3位双键质子信号;将得到的1hnmr图谱应用mestrenova9.0软件定标,仔细校正基线与相位,选择与样品中其它质子峰分离较好的特征峰作为待测成分的定量峰。对于甘油定量应考虑其在样品中与其他成分的重叠效应,甘油果糖氯化钠注射液中,甘油在δ3.80-3.75处的峰与样品中其他成分有重叠;参芎葡萄糖注射液中δ3.80-3.75及δ3.57-3.52处的峰与其他成分存在重叠。因此,为保证积分的一致性与准确性,对四种注射液的甘油统一选择δ3.657-3.608作为定量质子峰的积分区间;选择内标马来酸δ6.511-6.293为定量质子峰的积分区间;tsp其信号峰在δ0.00,用作定零点,排除内标物对待测成分定量峰的影响。
1.6方法学考察
专属性考察:精密称取甘油、1,2-丙二醇,加入超纯水、内标溶液,转移至5mm核磁管,加封后用于1h-nmr测试;精密称取甘油、1,3-丙二醇,加入超纯水、内标溶液,转移至5mm核磁管,加封后用于1h-nmr测试。结果见表3、图2。
线性关系考察:精密称取甘油对照品于容量瓶中,用内标溶液稀释,加入适量超纯水并定容至刻度,配置系列摩尔浓度的对照品溶液,分别取适量装入nmr管中,按“1.4”项下条件采集图谱,以定量峰与内标物峰积分面积之比为横坐标(x),以对照品浓度(mmol/l)为纵坐标(y),进行线性回归。
准确性考察:以已知甘油浓度的真实值为基准,测量值为参考,用相对误差百分率表示定量方法的准确度,相对误差率的绝对值越小,代表定量方法的准确度越高。采集50%浓度和100%浓度自制供试品溶液的1h-nmr图谱,采用内标定量峰面积法对甘油和内标物的定量峰分别进行积分,按照1h-nmr内标法根据积分结果依照下式计算甘油的含量,与实际浓度相比较:
其中,px为待测物的浓度(mmol/l),pma为内标马来酸的浓度(mmol/l),ax为待测物中定量峰面积,ama为内标马来酸的定量峰面积,nx为待测物的定量质子数,nma为内标马来酸定量质子数。结果见表4。
加样回收率试验:精密称取1.44mg乳酸钠,0.59mg甘油,按照1.2项下制备,作为空白对照溶液,共6份;在空白对照溶液中精密加入0.55mg甘油对照品,涡旋混匀,作为加样回收样品溶液,共6份,按照1.4项下采集样品,分别记录空白对照溶液,加样回收样品溶液的定量峰面积并计算其加样回收率。
精密度试验:取自制供试品溶液,按上述1h-nmr测试条件下连续测定6次,记录待测成分的定量峰面积,计算测试样品浓度及日内精密度的rsd。
稳定性试验:取同一自制供试品溶液分别在0,2,4,6,8,12h进样测定,记录积分面积,计算rsd值。
重复性试验:取自制供试品溶液,共6份,按上述1h-nmr测试条件进行测定,记录积分面积,计算rsd值。
表3专属性考察结果
以甘油积分面积均值x为横坐标,浓度y(mmol/l)为纵坐标,绘制甘油对照品标准曲线,计算出标准曲线的回归方程和相关系数。标准曲线方程为y=20.912x-0.4351,对照品在5.48~175.37mmol/l范围内的线性关系良好(r2=1)。
表4自制样品准确度结果(n=6)
注:绝对误差=测量值-真实值,相对误差率=绝对误差/真实值;
实验结果见下表5。
表5定量核磁法方法学考察
1.7定量核磁法测定不同种类注射剂中甘油的含量
取样品适量,精密称定,按照“1.3”项下方法制备供试品溶液,按“1.4”项下条件进行测定。记录样品nmr图谱,计算含量。结果见表6。
表6样品含量测定
实施例2
四种注射液13c-nmr标准指纹图谱的建立
1.1仪器与试药
brukeravanceⅲ600型超导核磁共振波谱仪,质子激发频率600.25mhz,配置bbfo正相观察宽带探头和bvt3200数字控温仪;5mm标准核磁样品管,美国norell公司生产;mettlertoledoxp6天平(瑞士mettler公司)。
d2o氘代度≥99.9%,美国sigma-aldrich公司;3-(三甲基甲硅烷)丙酸-d4钠盐(tsp)氘代度>98%,sigma-aldrich公司生产;甘油(glycerol,c3h8o3),≥99.5%,美国sigma-aldrich公司生产;马来酸(maleicacid,c4h4o4),≥99.94%,美国sigma-aldrich公司生产;乳酸钠(sodiumlactate,c3h5nao3),≥99.0%,美国sigma-aldrich公司生产;1,2-丙二醇(1,2-propanediol,c3h8o2),≥99.5%,美国sigma-aldrich公司生产;1,3-丙二醇(1,3-propanediol,c3h8o2),≥98%,美国sigma-aldrich公司生产。
四种注射液名称、生产厂家及生产批号分别为:甘油果糖注射液,购自南京正大天晴制药有限公司,生产批号1504242;参芎葡萄糖注射液,购自贵州景峰注射剂有限公司,生产批号20150682;消痔灵注射液,购自吉林省吉安市益盛药业股份有限公司,生产批号15050505;依托咪酯乳状注射液,购自江苏恩华药业股份有限公司,生产批号20150803。
1.2自制供试品溶液的制备
称取马来酸标准品于容量瓶中,用d2o(重水)溶解并定容至刻度线制得内标溶液,所述内标溶液中含有543.11mmol/l马来酸。
精密称取14.64mg乳酸钠标准品,12.05mg甘油,加入480μl超纯水,120μl内标溶液,得自制供试品溶液,将所述供试品溶液可转移至5mm核磁管中,直接用于13c-nmr测试。
1.3四种注射液供试样品溶液的制备
精密移取0.1ml消痔灵注射液,用0.3ml超纯水稀释,加入0.1ml内标溶液,得消痔灵供试品溶液;精密移取0.1ml甘油果糖氯化钠注射液,用0.3ml超纯水稀释,加入0.1ml内标溶液,0.3ml超纯水,得甘油果糖氯化钠供试品溶液;精密移取0.4ml参芎葡萄糖注射液,加入0.1ml内标溶液,得参芎葡萄糖供试品溶液;精密移取0.4ml依托咪酯乳状注射液,加入0.1ml内标溶液,得依托咪酯乳状供试品溶液;将上述供试品溶液可转移至5mm核磁管中,直接用于13c-nmr测试。
1.413c-nmr指纹图谱的测定
将核磁管置于核磁共振仪中,采用门控去耦序列测定注射液样品溶液的13c-nmr指纹图谱和自制品溶液的13c-nmr图谱,其中测试参数及条件为:门控去耦序列zgig,观测频率603.80mhz,测定温度298k,谱宽36057.7hz,90°脉冲宽度16.28μs,采样数据点16384,采样时间0.4544s,扫描次数32次,采样前弛豫延迟时间10s,接受增益2050。
1.5样品13c-nmr图谱分析
根据13c-nmr谱,鉴定甘油中共2组碳原子信号,包括δ73.1,2位碳原子信号和δ62.5,1、3位碳原子信号;马来酸共2组碳原子信号,包括δ132.6,2、3位双键碳原子信号和δ170.8,1、4位羰基碳原子信号;门控去耦是全去藕图谱,可以为碳原子定量提供信息,但不是待测组分中所有官能团上的碳原子都可以作为定量计算的选择。由于本次实验d1时间的限制,甘油与马来酸两种分子中不同碳原子t1的不同及门控去耦存在的部分noe效应,导致响应效果的差异。甘油中2组碳原子信号的峰强度(或面积)与其个数成正比,而马来酸δ171.77的碳原子与δ135.62的碳原子,由于其所处的环境不同,与氢核相近的碳原子的信号被增强,故马来酸δ135.62的峰强度(或峰面积)与碳原子数成正比,δ171.77的峰强度(或峰面积)与碳原子数不成比例。因此,选择内标马来酸δ135.62处的官能团上的碳原子为定量峰,甘油δ73.07处的碳原子作为定量峰,分别对峰面积和峰强度进行积分和记录。
1.6方法学考察
专属性考察:精密称取甘油、1,2-丙二醇,加入超纯水、内标溶液,转移至5mm核磁管,加封后用于13c-nmr测试;精密称取甘油、1,3-丙二醇,加入超纯水、内标溶液,转移至5mm核磁管,加封后用于13c-nmr测试。结果见表7、图3。
线性关系考察:精密称取甘油对照品于容量瓶中,用内标溶液稀释,加入适量超纯水并定容至刻度,配置系列摩尔浓度的对照品溶液,分别取适量装入nmr管中,按“1.4”项下条件采集图谱,以对照品甘油与内标马来酸的定量峰强度比为横坐标(x),以甘油与内标马来酸的摩尔浓度比为纵坐标(y)进行线性回归,建立13c-nmr峰强度-标准曲线方程。
准确性考察:以已知甘油浓度的真实值为基准,测量值为参考,用相对误差百分率表示定量方法的准确度,相对误差百分率的绝对值越小,代表定量方法的准确度越高。采集50%浓度和100%浓度自制供试品溶液的13c-nmr图谱,采用公式y=ax+b计算甘油的含量,与实际浓度相比较;
其中x代表定量成分定量峰的峰强度与内标定量峰的峰强度之比,y代表定量成分与内标的摩尔浓度比,a和b分别为标准曲线方程的系数。结果见表8。
加样回收率试验:精密称取12.20mg乳酸钠,5.02mg甘油,按照1.2项下制备,作为空白对照溶液,共6份;在空白对照溶液中精密加入5.05mg甘油对照品,涡旋混匀,作为加样回收样品溶液,共6份,按照1.4项下采集样品,分别记录空白对照溶液,加样回收样品溶液中待测成分的定量峰的强度,计算甘油的测定值,根据样品中甘油的原有量和测定量,加入量,计算自制样品中甘油的加样回收率。
精密度试验:取自制供试品溶液,按上述13c-nmr测试条件下连续测定6次,记录待测成分的定量峰强度,计算精密度的rsd。
稳定性试验:取同一自制供试品溶液分别在0,2,4,6,8,12h进样测定,记录待测成分的定量峰强度,计算rsd值。
重复性试验:取自制供试品溶液,共6份,按上述13c-nmr测试条件进行测定,记录待测成分的定量峰强度,计算rsd值。
表7专属性考察结果
以甘油对照品与马来酸峰强度比x为横坐标,以甘油与马来酸的摩尔浓度比y为纵坐标,建立标准曲线方程,并计算方程的相关系数,标准曲线方程为y=1.8132x+0.132,r2=0.9978,说明甘油对照品在27.17~869.58mmol/l范围内线性关系良好。
表8自制样品浓度的准确度考察结果(n=6);
注:绝对误差=测量值-真实值,相对误差率=绝对误差/真实值;
实验结果见下表9。
表9定量核磁法方法学考察
1.7定量核磁法测定不同种类注射剂中甘油的含量
取样品适量,精密称定,按照“1.3”项下方法制备供试品溶液,按“1.4”项下条件进行测定。记录样品cmr图谱,计算含量。结果见表10。
表10样品含量测定
上述参照具体实施方式对该一种注射剂中甘油含量的检测方法进行的详细描述,是说明性的而不是限定性的,可按照所限定范围列举出若干个实施例,因此在不脱离本发明总体构思下的变化和修改,应属本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!