一种测定饮用水中痕量双氯酚和六氯酚的气相色谱法的制作方法
本发明涉及有害痕量物质分析检测的方法,具体涉及到一种测定饮用水中痕量双氯酚和六氯酚的气相色谱法。
背景技术:
双氯酚(cas:97-23-4)和六氯酚(cas:70-30-4)是两种杀菌剂,常用于化妆品中用于杀菌,可微量溶解于水中并稳定持久地存在于水体中。这两类物质随着化妆品的广泛使用逐渐成为污染饮用水的有害化学物质来源。
当前对于饮用水中这两种物质痕量检测相关研究报道较少。有关于该两种物质痕量检测主要方法为色谱以及色谱-质谱联用法,前处理方法主要集中于采用液-液萃取和固相萃取两种方法。但是采用液-液萃取法存在需要使用大量有机溶剂的缺陷;而采用固相萃取法存在的缺陷有:一是可采用的固相萃取填料种类有限,商品化产品种类少,二是采用固相萃取柱模式吸附需要将大量的水以一定速度通过固相萃取柱,以使固相萃取柱中吸附剂可以吸附水中目标物,这一过程通常需要耗费大量的时间。
发明人在前期的研究中应用2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物吸附剂对水中该两种化合物进行了吸附实验,结果显示2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物吸附剂对目标物具有良好的吸附效果。在此基础上,发明人进一步对所研制的吸附剂富集水中目标化合物的性能和应用方法进行了优化,建立了以2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物为吸附剂的饮用水中痕量双氯酚和六氯酚检测的气相色谱法。
技术实现要素:
为了克服现有饮用水中痕量双氯酚和六氯酚检测前处理中存在的液-液萃取法需要使用大量有机溶剂的缺陷;以及固相萃取法可用固相萃取产品种类少,吸附过程需要耗费大量时间的不足,本发明要解决的技术问题在于提供一种基于新型吸附剂分散固相萃取快速吸附的、适合于饮用水中双氯酚和六氯酚检测的气相色谱法。
本发明是通过以下技术方案来实现上述目标的。
一种测定饮用水中痕量双氯酚和六氯酚的气相色谱法,其特征在于,包括如下步骤:
步骤1化合物的吸附:在分液漏斗中添加一定量的饮用水,向其中添加0.40g的2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物吸附剂,震荡一定时间使吸附剂吸附水中的痕量双氯酚和六氯酚;
步骤2化合物的脱吸附:将分液漏斗中的吸附剂与水混合物转移至具塞离心管中离心,使固体吸附剂与水溶液分离并弃去上清液,将所有下层固体吸附剂合并至一个离心管中,向其中添加一定量的盐酸溶液溶解固体吸附剂,完成被吸附化合物的脱吸附;
步骤3化合物的萃取与衍生化:向上述离心管中添加一定量无水硫酸钠及有机溶剂萃取,涡旋、离心,分取上清液至衍生瓶中,向其中添加衍生化试剂,密封、混匀后置于恒温水浴中完成衍生化过程;
步骤4化合物的分析测试:向衍生瓶中添加饱和碳酸氢钠溶液及一定量的碳酸氢钠固体粉末,涡旋并超声,吸取上层有机溶液过滤后应用气相色谱法进行分析测试。
其中,
步骤1中所述的饮用水量为200ml,震荡的时间为15min。
步骤2中所述的盐酸溶液为浓盐酸与水按照体积比1:1配制而成,用量为4.00ml。
步骤3中添加的无水硫酸钠为4.0g,有机萃取溶剂为5.00ml乙酸乙酯,衍生化试剂为0.10ml乙酸酐溶液,水浴温度为50℃,衍生化时间为30min。
步骤4中所述的饱和碳酸氢钠溶液为2.0ml和碳酸氢钠固体粉末为0.20g,涡旋时间为30s至60s,超声时间为2min,滤膜为有机相滤膜,孔径0.22μm。
上述步骤中涡旋为涡旋1min至2min,离心为以4500rpm转速运行3min。
用于测定的气相色谱法分析测试条件为:
a)色谱柱:hp-5毛细管柱,30m×0.32mm,0.25μm膜厚;恒流模式,柱流速:2.00ml/min。
b)进样口温度:250℃;进样方式:不分流进样;进样量:2μl;尾吹气流量:60ml/min。
c)升温程序:60℃(保持2min),以10℃/min速率升温至190℃(保持0min),以20℃/min速率升温至300℃(保持4.5min)。
d)检测器:电子俘获检测器,温度:325℃。
e)载气:高纯氮气(纯度≥99.999%)。
在方法研发的过程中,发明人根据吸附目标物的特性对吸附剂的用量、提取溶剂的选择与配比、目标物衍生化方法的选择及优化、色谱分离条件的选择和优化等多因数进行了考察与优化,在此基础上提出了相对较优的检测方法。同时,出于对目标物定量准确性考虑,本方法在无法获得目标物同位素从而进行同位素内标法定量的前提下采用基质校正曲线对目标物进行定量,以尽量消除系统误差提高定量的准确性。
本发明的优点在于:
(1)本发明所采用的新型吸附剂2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物可以采用分散固相萃取的方式对水中痕量双氯酚和六氯酚进行快速的吸附,相较于固相萃取的方式可以节省大量的吸附时间;
(2)本发明利用2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物吸附剂可溶解于酸的特点,应用盐酸溶液对吸附目标物后的吸附剂进行溶解,可以使得目标物从吸附剂上完全脱吸附;
(3)本发明仅适用少量的有机溶剂作为目标物的萃取溶剂,相较于液-液萃取法需要使用大量有机溶剂具有显著的安全、环保优点和经济优势。
附图说明
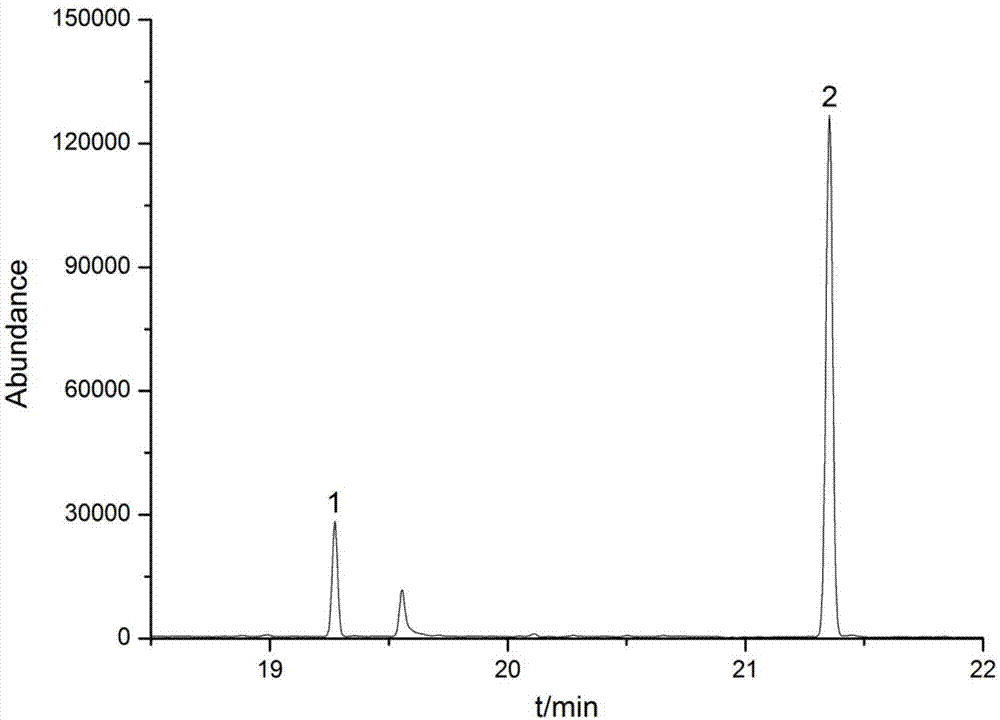
图1是具体实施方式中双氯酚和六氯酚的基质标准溶液色谱图,浓度为200.0μg/l,其中1是双氯酚、2是六氯酚。
具体实施方式
为进一步公开而不是限制本发明,以下结合实例对本发明作进一步的详细说明。
(1)本发明实施例中所涉及到的试剂药品如下:
双氯酚和六氯酚固体标准品,纯度大于等于98.0%,上海阿拉丁生化科技股份有限公司;
乙酸酐,乙酸乙酯,无水硫酸钠、碳酸氢钠,分析纯,国药集团;
盐酸,优级纯,国药集团;水为符合gb/t6682规定的一级水。
(2)本发明实施例中所涉及到的仪器如下:
kh-75a型电热恒温鼓风干燥箱,广州市康恒仪器有限公司;
7890b型气相色谱仪,配电子俘获检测器,美国安捷伦科技有限公司。
(3)气相色谱仪分析测试条件:
a)色谱柱:hp-5毛细管柱,30m×0.32mm,0.25μm膜厚;恒流模式,柱流速:2.00ml/min。
b)进样口温度:250℃;进样方式:不分流进样;进样量:2μl;尾吹气流量:60ml/min。
c)升温程序:60℃(保持2min),以10℃/min速率升温至190℃(保持0min),以20℃/min速率升温至300℃(保持4.5min)。
d)检测器:电子俘获检测器,温度:325℃。
e)载气:高纯氮气(纯度≥99.999%)。
(4)基质校正曲线的制作和检出限、定量限的确定
准确称量上述两种化合物并以甲醇溶解定容,配制成浓度为1000mg/l的标准储备液,-4℃下保存。使用时将标准储备液以去离子水逐步稀释,配制成浓度梯度为10.0μg/l、20.0μg/l、40.0μg/l、100.0μg/l和200.0μg/l的标准使用液。
取五个500ml的分液漏斗并向其中各添加200ml去离子水,分别取上述标准使用液各5.00ml,之后向各漏斗中添加0.40g的2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物吸附剂,震荡15min使吸附剂吸附;将分液漏斗中的水添加至50ml具塞离心管中以4500rpm转速离心3min,使固体吸附剂与水溶液分离并弃去上清液,将所有下层固体吸附剂合并至一个离心管中,向其中添加4.00ml稀释1倍的盐酸溶液,待固体吸附剂溶解完成后实现被吸附目标物的脱吸附;向上述离心管中添加4.0g无水硫酸钠,再添加5.0ml乙酸乙酯并涡旋1min、4500rpm转速离心3min,取上清液至衍生瓶中,并向其中添加0.10ml乙酸酐溶液,密闭、混匀后静置于50℃的水浴中衍生化30min,完成酯化衍生化过程;向上述衍生瓶中添加上述溶液中添加2.0ml饱和碳酸氢钠溶液和0.20g的碳酸氢钠固体粉末,涡旋时间为30s,超声时间为2min,吸取上层有机溶液过孔径为0.22μm的有机相滤膜,之后以气相色谱法分析测试。
以双氯酚和六氯酚在样品溶液的浓度为x轴,双氯酚和六氯酚在气相色谱仪上的色谱峰峰面积为y轴绘制基质标准曲线并用于外标法定量。
以信噪比s/n的三倍值为方法的检测限(lod,lod=3s/n),以信噪比s/n的十倍为方法的定量限(loq,loq=10s/n),并结合添加基质的体积计算各个化合物在水中的检出限和定量限。
上述基质标准曲线相关参数、lod和loq相关信息见表1。
表1.双氯酚和六氯酚基质标准曲线、检测限和定量限相关信息
(5)2-萘酚-6-磺酸根-镁铝型水滑石吸附剂的合成
为了是本领域的技术人员能够重复实现本发明的相关实验,现在提供一种本发明中使用的关键物质2-萘酚-6-磺酸根-镁铝型水滑石吸附剂的合成方法,如下:
①合成吸附剂所涉及到的试剂药品如下:
2-萘酚-6-磺酸钠,分析纯,国药集团;
mg6al2(oh)16co3·4h2o,分析纯,美国aldrich公司。
②合成吸附剂所涉及到的仪器如下:
excel型微波消解仪,上海屹尧仪器科技发展有限公司,消解罐体积为100ml;微波马弗炉(烧结炉),美国cem公司;vd53型真空干燥箱,德国宾德科技公司;hj-5多功能恒温搅拌器,金坛荣华仪器制造有限公司;fs-12型分液漏斗振荡器,日本新光科技有限公司;3k-15型离心机,德国sigma科技公司;bf518945c-1型箱式电阻炉(马弗炉),美国赛默飞世尔科技公司。
③合成吸附剂的具体步骤如下:
(a)第一次焙烧:将购买的镁铝型水滑石mg6al2(oh)16co3·4h2o置于马弗炉中,以升温速率5℃/min加热至温度为500℃,焙烧6h,得到焙烧产物mg6al2o8(oh)2;
(b)称量:于微波消解罐中称取9.870g插层剂2-萘酚-6-磺酸钠和7.236g的焙烧产物mg6al2o8(oh)2。
(c)微波晶化水热合成:将去离子水煮沸并保持30min,之后添加100ml至上述装有插层剂和焙烧产物的微波消解罐中,密闭,将微波消解罐置于微波消解仪中,在150℃下微波加热30min完成合成;
(d)洗涤与干燥:将微波罐中的全部固体与液体倒出,以煮沸30min以上的去除二氧化碳的去离子水经加热搅拌、振荡洗涤并离心,90℃下真空干燥12h,研磨。
(e)第二次焙烧:将上述合成的材料置于马弗炉中,以升温速率5℃/min加热至温度为500℃,焙烧6h,得到用作吸附剂的焙烧产物,研磨密封保存。
实施例1
本实施例1采用自来水为样品基质进行加标回收实验以验证本发明方法的可行性,并按照下列步骤进行处理:
1.化合物的吸附:
在500ml的分液漏斗中分别添加200ml的自来水,并向其中各添加5.00ml浓度分别为10.0μg/l、20.0μg/l和200.0μg/l的两种化合物标准溶液,制备得到三水平六平行的加标样品;再分别添加0.40g的2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物吸附剂,震荡15min使吸附剂吸附;
2.化合物的脱吸附:
将分液漏斗中的水添加至50ml具塞离心管中以4500rpm转速离心3min,使固体吸附剂与水溶液分离并弃去上清液,将所有下层固体吸附剂合并至一个离心管中,向其中添加4.00ml稀释1倍的盐酸溶液,待固体吸附剂溶解完成后实现被吸附化合物的脱吸附;
3.化合物的萃取与衍生化:
向上述离心管中添加4.0g无水硫酸钠,再添加5.00ml乙酸乙酯并涡旋1min、4500rpm转速离心3min,取上清液至衍生瓶中,并向其中添加0.10ml乙酸酐溶液,密闭、混匀后静置于50℃的水浴中衍生化30min,完成衍生化过程;
4、分析测试验证:
向上述衍生瓶中添加上述溶液中添加2.0ml饱和碳酸氢钠溶液和0.2g碳酸氢钠固体粉末,涡旋时间为30s,超声时间为2min,吸取上层有机溶液过孔径为0.22μm的有机相滤膜,之后以气相色谱法分析测试。
本实施例1的加标回收率实验相关参数见表2。
表2自来水样品的添加浓度及回收率实验数据(n=6)
实施例2
本实施例2采用井水为样品基质进行加标回收实验以验证本发明方法的可行性,按照下列步骤进行处理:
1.化合物的吸附:
在500ml的分液漏斗中分别添加200ml的井水,并向其中各添加5.00ml浓度分别为10.0μg/l、20.0μg/l和200.0μg/l的两种化合物标准溶液,制备得到三水平六平行的加标样品;再分别添加0.40g的2-萘酚-6-磺酸根-镁铝型水滑石焙烧产物吸附剂,震荡15min使吸附剂吸附;
2.化合物的脱吸附:
将分液漏斗中的水添加至50ml具塞离心管中以4500rpm转速离心3min,使固体吸附剂与水溶液分离并弃去上清液,将所有下层固体吸附剂合并至一个离心管中,向其中添加4.00ml稀释1倍的盐酸溶液,待固体吸附剂溶解完成后实现被吸附化合物的脱吸附;
3.化合物的萃取与衍生化:
向上述离心管中添加4.0g无水硫酸钠,再添加5.00ml乙酸乙酯并涡旋1min、4500rpm转速离心3min,取上清液至衍生瓶中,并向其中添加0.10ml乙酸酐溶液,密闭、混匀后静置于50℃的水浴中衍生化30min,完成衍生化过程;
4、分析测试验证:
向上述衍生瓶中添加上述溶液中添加2.0ml饱和碳酸氢钠溶液和0.2g碳酸氢钠固体粉末,涡旋时间为30s,超声时间为2min,吸取上层有机溶液过孔径为0.22μm的有机相滤膜,之后以气相色谱法分析测试。
本实施例2的加标回收率实验相关参数见表3。
表3井水样品的添加浓度及回收率实验数据(n=6)
以上所述实施例仅表达了本发明的实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员而言,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些均属于本发明的保护范围,因此本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!