一种测定叶黄素成分的方法与流程
本发明涉及分析化学技术领域,特别涉及一种一种测定叶黄素成分的方法。
背景技术:
叶黄素(lutein)是一种属于类胡萝卜素的黄色物质,广泛存在自然界中,在人体内,存在于血浆和眼睛的黄斑区,能够大量吸收蓝光,避免视网膜的光氧化损伤,同时作为一种抗氧化剂,能够清除自由基,保护视神经免受自由基的损害,因此被广泛应用于各种保健食品、食品及药品中。叶黄素的分子结构因具有多个共轭双键结构,易受光、氧、高温等因素的影响,在理论上可以存在多个异构体,包括顺式异构及其同分异构玉米黄质等。
在生物体内,不同异构体的生物活性不同,反式构象的生物活性也较顺式构象高很多,导致其生物学功能或效价差异显著,一旦叶黄素在制备和应用的过程中受到光照、加热、氧气等因素的影响,则不可避免地影响以叶黄素为主要活性成分的产品的质量。目前很少有人监测在叶黄素提取、制剂过程中顺式异构体的变化,特别是以叶黄素为原料的保健食品及食品普遍以总叶黄素含量的高低来评价产品的质量,使得产品的质量难以得到保障。现有的国家标准以纯度为总叶黄素含量的对照品,以反式叶黄素及其顺式异构体及同分异构体的总和作为定量指标,并且反式叶黄素及其顺式异构体及同分异构体未得到理想的分离,见图1,在其异构化的标准溶液的色谱图中,只可见三个色谱峰,并未见理论上存在或有关文献的多个顺式异构体及同分异构体,使得现有方法对叶黄素质量监控有很大的局限性。不仅如此,标准中该供试品制备方法相当繁琐,因而增加了叶黄素质量控制的成本。
技术实现要素:
有鉴于此,本发明旨在提出一种以叶黄素为原料的保健食品、食品、药品及叶黄素原料中全反式叶黄素及异构体的定性或定量方法。具体地,本发明提出了一种测定叶黄素成分的方法,该方法采用高效液相色谱检测,其中,色谱条件包括:以ymccarotenoidc30为色谱柱;以甲醇/水为流动相a,甲基叔丁基醚为流动相b;洗脱程序为0min~30min、0%~50%b,30min~40min、50%b;所述甲醇/水的比例为88~100:12~0。
优选地,甲醇/水的比例为92~100:8~0;更优选地,甲醇/水的比例为95:5。
进一步地,所述流动相a和/或所述流动相b中包含2,6-二叔丁基对甲酚。
优选地,所述2,6-二叔丁基对甲酚的浓度为0.1%。
进一步地,所述色谱条件包括:流速为0.7ml/min~1.1ml/min;柱温25℃~40℃。
进一步地,前述测定叶黄素成分的方法中,色谱条件包括:以规格为250mm×4.6mm,5μm的ymccarotenoidc30为色谱柱;以含0.1%的2,6-二叔丁基对甲酚、比例为95:5的甲醇/水溶液为流动相a,以甲基叔丁基醚为流动相b,洗脱程序为0min~30min、0%~50%b,30min~40min、50%b;流速为0.8ml/min,柱温为30℃,检测波长为445nm。
可选地,如前所述叶黄素成分包括但不限于全反式叶黄素。
本发明还提出一种检测叶黄素原料(如:叶黄素微粒)或含叶黄素的样品的方法,其特征在于,该方法包括:
供试品溶液的制备:将叶黄素微粒或含叶黄素的样品研细,取粉末置于容量瓶中,加入水,于室温下超声处理,再加入含bht的无水乙醇,超声处理,冷却至室温,定容;
上述供试品溶液用前述任一一种测定叶黄素成分的方法进行检测。
具体地,供试品溶液的制备包括:取叶黄素微粒或含叶黄素的样品,研细,取粉末约300mg,精密称定,置于100ml棕色容量瓶中,加入水5ml,于室温下超声处理20min,再加入含0.1%bht的无水乙醇至接近刻度,超声处理5min,冷却至室温,用含0.1%bht的无水乙醇定容至刻度,摇匀,离心,即得。(叶黄素微囊需再精密量取1ml至10ml棕色容量瓶中,0.1%bht的无水乙醇定容至刻度)。
更进一步地,前述检测叶黄素微粒或含叶黄素的样品的方法包括:
对照品溶液的制备:取叶黄素对照品,加含bht的无水乙醇制成含叶黄素的储备液,利用该储备液配制各浓度的对照品溶液;
供试品溶液的制备:将叶黄素微粒或含叶黄素的样品研细,取粉末置于容量瓶中,加入水,于室温下超声处理,再加入含bht的无水乙醇,超声处理,冷却至室温,定容;
上述对照品溶液和供试品溶液用权利要求1-8任一所述方法进行检测。据此可建立对照品的标准曲线,从而根据供试品检测结果计算有效成分的含量。
更具体地,所述对照品溶液可以按如下方式配制:取叶黄素对照品适量,精密称定,加入含0.1%bht(2,6-二叔丁基对甲酚)的无水乙醇制得每1ml含叶黄素100μg的对照品储备液;分别精密量取该储备液0.2、0.4、0.8、1.2、1.6、2.0ml,加入0.1%bht的无水乙醇稀释成浓度分别为2、4、8、12、16、20μg/ml的系列浓度的对照品溶液。
本发明提供的叶黄素各成分的定性或定量的分析方法,具有如下优点:
1.本方法包括以高活性成分全反式叶黄素为检测指标,可以更真实有效地评价叶黄素类产品的质量。
2.本方法的色谱条件可以实现全反式叶黄素及其主要的顺式异构体及同分异构体的有效分离,而且还能对全反式叶黄素进行准确定量。
3.本方法通过对照品对比、化合物的紫外光谱特性等对叶黄素及各异构体进行定性,检出多个化合物。
4.供试品处理方法采用水和无水乙醇提取的方式,操作简便,溶剂经济绿色安全。
5.本方法经过系统的方法学验证,本法简单快捷,专属性好,结果准确,重复性好,具有一定的推广价值。
附图说明
图1为国家标准中叶黄素对照品溶液异构化的hplc图谱;
图2为本发明的流动相程序ⅰ的供试品溶液液hplc图谱;
图3为本发明的流动相程序ⅱ的供试品溶液液hplc图谱;
图4为本发明的流动相程序ⅲ的供试品溶液液hplc图谱;
图5为本发明的流动相程序ⅳ的供试品溶液液hplc图谱;
图6为本发明的流动相程序ⅴ的供试品溶液液hplc图谱;
图7为本发明的流动相甲醇/水=86:14的供试品溶液液hplc图谱;
图8为本发明的流动相甲醇/水=88:12的供试品溶液液hplc图谱;
图9为本发明的流动相甲醇/水=90:10的供试品溶液液hplc图谱;
图10为本发明的流动相甲醇/水=92:8的供试品溶液液hplc图谱;
图11为本发明的流动相甲醇/水=95:5的供试品溶液液hplc图谱;
图12为本发明的流动相甲醇/水=100:0的供试品溶液液hplc图谱;
图13为本发明的流速为0.8ml/min的供试品溶液液hplc图谱;
图14为本发明的柱温为30℃的供试品溶液液hplc图谱;
图15为本发明的反式叶黄素纯度验证结果;
图16为本发明的反式叶黄素对照品溶液hplc图谱;
图17为本发明的反式玉米黄质对照品溶液hplc图谱;
图18为本发明的叶黄素异构化对照品溶液的hplc图谱;
图19为本发明的供试品溶液的hplc图谱;
图20为本发明的峰1的紫外光谱图;
图21为本发明的峰2的紫外光谱图;
图22为本发明的峰3的紫外光谱图;
图23为本发明的峰4的紫外光谱图;
图24为本发明的峰5的紫外光谱图;
图25为本发明的峰6的紫外光谱图;
图26为本发明的峰7的紫外光谱图;
图27为本发明的峰8的紫外光谱图;
图28为本发明的峰9的紫外光谱图;
图29为本发明的峰10的紫外光谱图;
图30为本发明的峰11的紫外光谱图;
图31为本发明的峰12的紫外光谱图;
图32为本发明的专属性实验反式叶黄素对照品溶液的hplc图;
图33为本发明的专属性实验阴性供试品溶液的hplc图;
图34为本发明的专属性实验供试品溶液的hplc图。
具体实施方式
鉴于全反式叶黄素的优异活性,本发明旨在提出一种以叶黄素为原料的保健食品、食品、药品及叶黄素微粒中全反式叶黄素及异构体的定性或定量方法,本领域技术人员可以借鉴本文内容,适当改进参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明提供的测定方法中所用药品、试剂、仪器,未注明生产厂商者,均为可以通过市购获得的常规产品。未注明具体实验条件者,按照常规条件或制造商建议的条件进行。
仪器:高效液相色谱仪(dionexultimate3000、agilent1260、shimadzulc-20at,配备dad检测器或者uv检测器);电子分析天平(mettlerxp6、mettlertoledoms105du、sartoriusbsa224s-cw);台式高速离心机(h1650-w、湖南湘仪实验室仪器开发有限公司);数控超声波清洗器(kq-500db,昆山市超声仪器有限公司);超纯水仪(milli-q,美国密理博公司);光照稳定性实验箱(ich-110型稳定性试验箱memmert,德国)。
试剂:甲醇(色谱纯,美国迈瑞达公司、美国默克公司);无水乙醇(色谱纯,美国迈瑞达公司);甲基叔丁基醚(色谱纯,美国迈瑞达公司);乙腈(色谱纯,美国天地公司);无水乙醇(分析纯,南京化学试剂有限公司);dmf(分析纯,北京百灵威科技有限公司);水(超纯水,自制);2,6-二叔丁基对甲酚(bht,化学纯cp,国药集团化学试剂有限公司);碘(分析纯,国药集团化学试剂有限公司)
对照品及样品:叶黄素对照品(即反式叶黄素)(含量以85.6%计,批号:lotlrab3708,购自sigma-aldrich公司);玉米黄质对照品(即反式玉米黄质)(ch13190320,成都克洛玛生物科技有限公司);叶黄素微粒(2017120607、2017120608、2018010602批)、浙江医药股份有限公司);叶黄素微粒阴性样品(由辛烯基琥珀酸淀粉钠、白砂糖、食用玉米淀粉等辅料混合制备而成),美灵片样品(20190101、20190102、20190103批),由江苏康缘药业股份有限公司健康产品部门提供
实施例1色谱条件的选择
对照品溶液的制备:取叶黄素对照品适量,精密称定,加含0.1%bht的无水乙醇制成每1ml含叶黄素100μg的溶液,即得对照品储备液。分别精密量取该储备液0.2、0.4、0.8、1.2、1.6、2.0ml于10ml量瓶中,加0.1%bht的无水乙醇稀释成浓度分别为2、4、8、12、16、20μg/ml的系列浓度的对照品溶液。
供试品溶液的制备:取叶黄素微粒(2017120608批)约300mg,精密称定,置于100ml棕色容量瓶中,加水5ml,于室温下超声(功率100w)处理20min,再加入含0.1%bht的无水乙醇至接近刻度,超声(功率100w)处理5min,冷却至室温,用0.1%bht的无水乙醇定容至刻度,再精密量取1ml至10ml棕色容量瓶中,用0.1%bht的无水乙醇稀释至刻度,摇匀,离心,即得。
1波长的选择
采用dad检测器,在200nm~500nm范围内对叶黄素对照品溶液进行全波长扫描,结果显示叶黄素的最大吸收波长在444nm处,为与国家标准中的色谱条件保持一致,故选择445nm作为检测波长。
2流动相的选择
2.1流动相比例的选择
首先参照国家标准中的色谱条件,即以ymccarotenoidc30为色谱柱,柱温为30℃,流速为1.0ml/min,检测波长为445nm,以甲醇/水(88:12)为流动相a,甲基叔丁基醚为流动相b,两相均含有0.1%bht,梯度洗脱,程序ⅰ为0~18min、0%~90%b,18~18.1min,90%~0%b,18.1~28min,0%b,结果见图2。由图可见,采用国家标准中的条件反式叶黄素峰7与峰6基本重合,与峰8的分离度约1.45,也未达到基线分离,因此首先考虑调整a、b两相的比例,考察不同的梯度洗脱程序下反式叶黄素与各异构体之间的分离效果。进一步设置的梯度程序如下:梯度程序ⅱ为0~20min、0%→70%b,20~20.1min、70%~0%b,20.1~30min、0%b;程序ⅲ为0~25min、0%~60%b,25~25.1min,60%~0%b,25.1~35min、0%b;程序ⅳ为0~30min,0%~50%b,30~30.1min、50%~0%b,30.1~40min、0%b;程序ⅴ为0~35min、0%~40%b、35~45min,40%b。结果见图3~6,从图可以看出随着a相比例的增大,反式叶黄素峰7与相邻异构体峰6和峰8的分离度有所增大,峰7与峰8在程序ⅲ中可达到基线分离,但是峰7与峰6不管在哪个程序下始终无法达到基线分离,且保留时间由16min延长为32min。由此可见改变a、b两相的比例,对于改善峰7与峰6的分离效果虽然有一定的作用,但效果并非十分明显。相较而言,程序ⅳ作为流动相梯度,分离效果稍好,保留时间约25min,峰7与峰6的分离度能达到1.40-1.50,已具备一定的分离效果。
2.2流动相甲醇/水比例的选择
以ymccarotenoidc30为色谱柱,柱温为30℃,流速为1.0ml/min,检测波长为445nm,以甲醇/水为流动相a,甲基叔丁基醚为流动相b,两相均含有0.1%bht,梯度洗脱0min~30min、0%~50%b,30min~40min、50%b,在此条件下主要考察a相甲醇/水的比例分别为86:14、88:12、90:10、92:8、95:5和100:0时反式叶黄素峰7与相邻异构体峰6与峰8的分离效果,结果见图7~12。由图可见,随着甲醇比例的增高,分离度逐渐增大,峰7与峰8的分离度由1.5增加为5.0,与峰6的分离度由1.0增加为2.5,且保留时间由28min减少为14min,甲醇/水95:5至100:0峰7与相邻异构体的峰均可得达到基线分离,甲醇/水的比例为92~100:8~0已经具有较好的分离效果,综合考虑其他峰的分离及溶剂的经济性优先选择甲醇/水的比例为95:5,此条件下分离度约1.9,保留时间约22min。此外,国标中a、b两相均含有0.1%bht,配制繁琐,因此后续过程只在a相或者b相含0.1%bht,发现在本发明的条件下,并不影响分离效果,且具体浓度可以根据实际情况调整。
3不同流速的考察
以ymccarotenoidc30为色谱柱,以甲醇/水(95:5,含0.1%bht)为流动相a,甲基叔丁基醚为流动相b,梯度洗脱,0min~30min、0%~50%b,30min~40min、50%b,柱温为30℃,检测波长为445nm,分别考察流速为0.7、0.8、0.9、1.0、1.1ml/min对反式叶黄素与其异构体的分离效果及含量的影响,结果显示反式叶黄素在不同流速下与相邻异构体均可达到基线分离,且含量无明显差异,说明该方法在流速为0.7ml/min~1.1ml/min范围内耐用性良好。综合考虑分离度、保留时间并且兼顾其余异构体的分离,优先选择流速0.8ml/min,如图13所示,该条件下保留时间为23.6min,分离度为1.93,对称因子为1.06,理论板数为79684。
4不同柱温的考察
在上述条件下,分别考察柱温为25℃、30℃、35℃、40℃时对反式叶黄素及与相邻异构体的分离效果及含量的影响,结果显示反式叶黄素在不同柱温下与相邻异构体均有较好的分离效果,且含量无明显性差异,说明该方法在柱温25℃~40℃范围内耐用性良好,优先选择常用温度30℃,如图14所示,该条件下保留时间为23.5min,分离度为1.92,对称因子为1.06,理论板数为82472。
5不同仪器的考察
在上述条件下,分别考察岛津(shimadzu-20at)、安捷伦(agilent-1260)、戴安(dionexultimate3000)三个品牌的仪器对反式叶黄素与其异构体的分离效果及含量的影响,结果采用三个品牌的仪器,叶黄素均可得到较好的分离,并且含量无明显差异,说明仪器耐用性较好。
6色谱峰纯度验证
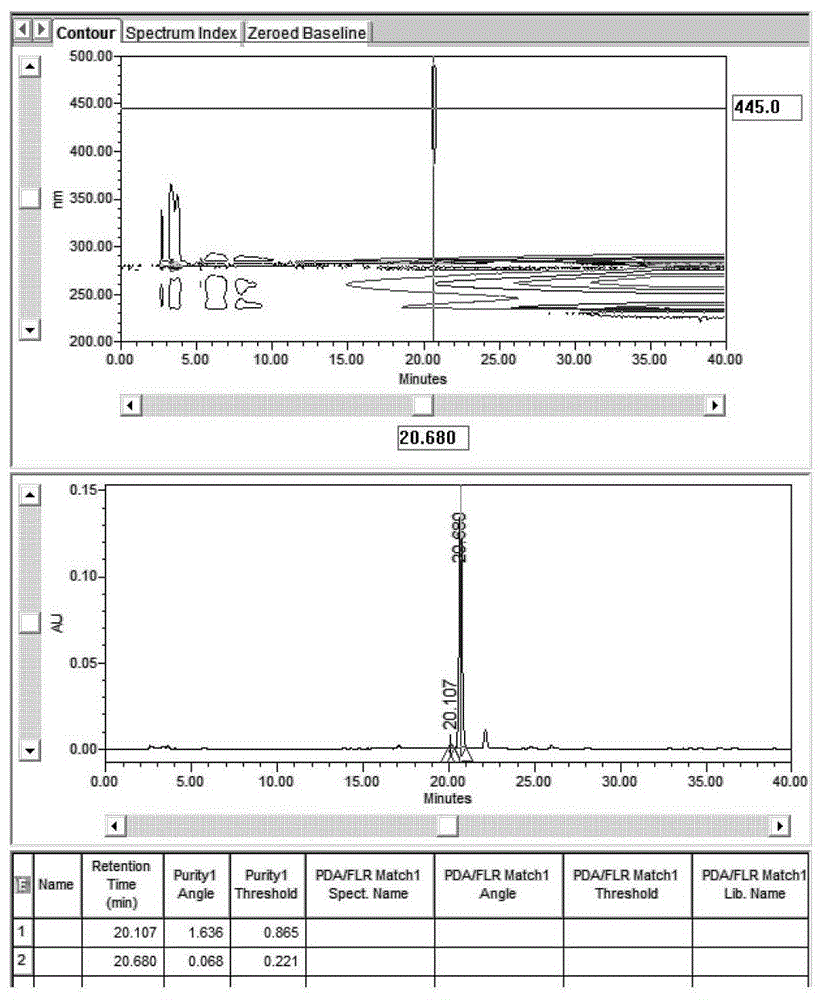
通过以上考察得到叶黄素的色谱条件为:色谱柱为ymccarotenoidc30(250mm×4.6mm,5μm);以甲醇/水(95:5,含0.1%bht)为流动相a,以甲基叔丁基醚为流动相b,梯度洗脱,0min~30min、0%~50%b,30min~40min、50%b;流速为0.8ml/min;柱温30℃;检测波为445nm;进样量10ul。在此条件下采用dad检测器对供试品色谱中反式叶黄素峰7进行峰纯度验证,结果如图15所示,从结果可以看出纯度角小于纯度阈值,叶黄素色谱峰纯度符合要求。
实施例2供试品制备方法的选择
首先采用2.1国标的方法制备供试品溶液,结果检测不出叶黄素的峰,推测可能为叶黄素微粒为微囊包合物,导致采用国标的低极性有机溶剂无法提取出叶黄素的成分,因此,考虑采用合适的溶剂破坏微囊,使其中的叶黄素得以释放,再采用无水乙醇进行提取,在此思路下对提取条件进行优化。
1破膜溶剂的选择
考察水、dmf、无水乙醇作为破膜溶剂对全反式叶黄素含量的影响。取叶黄素微粒(2017120608批)约300mg,精密称定,置于100ml棕色容量瓶中,分别加入水、dmf、无水乙醇各5ml,于室温下超声(功率100w)处理20min,再加入含0.1%bht的无水乙醇至接近刻度,超声(功率100w)处理5min,冷却至室温,用0.1%bht的无水乙醇定容至刻度,再精密量取1ml至10ml棕色容量瓶中,0.1%bht的无水乙醇稀释至刻度,摇匀,离心,即得。结果见表1。结果显示,以水作为破膜溶剂时,全反式叶黄素含量明显高于dmf、无水乙醇,且成本低廉。因此,优选用水作为破膜溶剂。
表1不同破膜溶剂对全反式叶黄素含量的影响
2破膜温度的选择
考察超声温度分别为20℃、30℃、40℃、60℃对全反式叶黄素含量的影响,结果见表2。结果显示,不同破膜温度对全反式叶黄素含量无明显差异,但是随着温度的升高,含量有下降趋势,综合考虑温度对叶黄素的敏感性以及操作的方便性,优选室温作为超声温度。
表2破膜温度对全反式叶黄素含量的影响
3破膜溶剂量的选择
考察破膜溶剂水的体积分别为2ml、5ml、7.5ml、10ml对全反式叶黄素含量的影响,结果见表3。结果显示,不同破膜溶剂量对全反式叶黄素含量无明显差异,为保证破膜充分以及全反式叶黄素的溶解度,最终选择5ml作为破膜溶剂量。
表3不同破膜溶剂量对叶黄素含量的影响
4破膜时间的选择
考察破膜时间5min、10min、20min、30min对全反式叶黄素含量的影响,结果见表4,结果显示,不同破膜时间对全反式叶黄素含量无明显影响,但破膜10min、20min的含量相对较高,为了保证破膜充分,最终选择20min作为破膜时间。
表4不同破膜时间对叶黄素含量的影响
5无水乙醇提取时间的选择
考察无水乙醇提取时间0min、5min、10min、20min为对全反式叶黄素含量的影响,结果见表5。结果显示,无水乙醇的不同提取时间对叶黄素含量无明显影响。考虑到超声有助于叶黄素分散均匀以及节约时间,最终选择5min作为无水乙醇提取时间。
表5无水乙醇提取时间对叶黄素含量的影响
通过以上考察,得到供试品溶液制备方法为:取叶黄素微粒约300mg,精密称定,置于100ml棕色容量瓶中,加水5ml,于室温下超声(功率100w)处理20min,再加入含0.1%bht的无水乙醇至接近刻度,超声(功率100w)处理5min,冷却至室温,用0.1%bht的无水乙醇定容至刻度,再精密量取1ml至10ml棕色容量瓶中,0.1%bht的无水乙醇稀释至刻度,摇匀,离心,即得。
实施例3叶黄素相关化合物的定性研究
1叶黄素对照品溶液的制备
取叶黄素(即反式叶黄素)对照品约5mg,精密称定,置于50ml棕色量瓶中,加入含0.1%bht的无水乙醇制得每1ml含叶黄素100μg的对照品储备液,再精密量取该储备液1.2ml置于10ml棕色量瓶中,0.1%bht的无水乙醇稀释制得浓度为12μg/ml的对照品溶液。
2玉米黄质对照品溶液的制备
取玉米黄质(即反式玉米黄质)对照品约5mg,精密称定,置于50ml棕色量瓶中,加入含0.1%bht的无水乙醇制得每1ml含叶黄素100μg的对照品储备液,再精密量取该储备液1.0ml置于50ml棕色量瓶中,0.1%bht的无水乙醇稀释制得浓度为2μg/ml的对照品溶液。
3叶黄素异构化对照品溶液的制备
取叶黄素对照品约5mg,精密称定于50ml透明容量瓶中,加入无水乙醇制得每1ml含叶黄素100μg的对照品储备液,取该储备液1ml置10ml透明量瓶中,加入0.1ml碘的乙醇溶液,用无水乙醇稀释至刻度,置于光照箱中放置1h,即得叶黄素异构化的对照品混合溶液。
4供试品溶液的制备
按照实施例2中所优化的供试品溶液的制备方法制备供试品溶液。
5测定
分别精密吸取上述各对照品溶液及供试品溶液,采用带有dad检测器的hplc进行测定。
6结果
叶黄素对照品、玉米黄质对照品、叶黄素异构化对照品溶液及供试品溶液的hplc图谱见图16~19(附图为局部放大图),各化合物的dad全波扫描光谱见图20~31。异构体可通过紫外光谱特性进行初步鉴定:顺式异构体与全反式叶黄素相比,单顺式异构体的最大吸收波长通常有4~6nm的蓝移,双顺式异构体则有8~12nm的蓝移;其次,单顺式异构体在330~340nm间有顺式吸收,且顺式双键越靠近分子的中心,其顺式吸收越大(通常用q值表示顺式吸收峰的强度);最后,叶黄素和b-胡萝卜素均为类胡萝卜素,都具有共同的异戊二烯结构,故他们相应位置异构体的洗脱顺序具有一致性据此。因此,根据对照品比对、紫外光谱特性及洗脱顺序,确定9个化合物结构并推测3个文献未见报告的化合物,各峰紫外吸收波长、q值、峰面积等参数见表6,各化合物结构如图32所示。具体如下:
峰7:首先与叶黄素对照品溶液的hplc保留时间一致,可初步判断为全反式叶黄素;其次吸收峰测定值420nm,444nm,472nm与已报道的423nm,444nm,472nm高度一致;另外q值测定值0.06与报道的0.05高度一致。因此综合判断峰7为化合物全反式叶黄素,结构见e。
峰8:首先与玉米黄质对照品溶液的hplc保留时间一致,可初步判断为全反式玉米黄质;其次吸收峰测定值335nm,425nm,450nm,478nm,与报道的427nm,450nm,477nm高度一致;另外q值测定值0.05,与报道的0.06基本一致。因此综合判断峰8为化合物全反式玉米黄质,结构见f。
峰3:首先吸收峰的测定值为330nm,412nm,438nm,464nm,其中最大吸收峰438nm与反式叶黄素的最大吸收峰444nm相比,发生了6nm的蓝移,根据文献报道单顺式异构体的最大吸收波长通常有4~6nm的蓝移,因此推断该化合物为单顺式叶黄素;其次四个吸收峰与报道的330nm,412nm,436nm,462nm高度一致;另外q值测定值0.47与报道的0.46高度一致;最后,参照胡萝卜素异构体的洗脱顺序,综合判断峰3为化合物13-顺式叶黄素,结构见a。
峰5:首先吸收峰的测定值为330nm,414nm,438nm,466nm,其中最大吸收峰438nm于与反式叶黄素的最大吸收峰444nm相比,发生了6nm的蓝移,而单顺式异构体的最大吸收波长通常有4~6nm的蓝移,因此推断该化合物为单顺式叶黄素;其次四个吸收峰与报道的330nm,414nm,438nm,464nm高度一致;另外q值测定值0.49与报道的0.45高度一致;最后,参照胡萝卜素异构体的洗脱顺序,综合判断峰5为化合物13’-顺式叶黄素,结构见b。
峰6:首先吸收峰测定值330nm,420nm,444nm,474nm,其中最大吸收峰444nm与反式玉米黄质的最大吸收峰450nm相比,发生了6nm的蓝移,由于单顺式异构体有4~6nm的蓝移,初步判断为单顺式玉米黄质;其次四个吸收峰与报道的331nm,444nm,473nm及报道的338nm,444nm,470nm高度一致;另外q值测定值0.10与报报道的0.43及报道的0.49的基本一致;最后参照胡萝卜素异构体的洗脱顺序,综合判断峰6为13或13’-顺式玉米黄质。结构见c和d。
峰9:首先吸收峰的测定值332nm,420nm,438nm,468nm,其中最大吸收峰438于与反式叶黄素的最大吸收峰444nm相比,发生了6nm的蓝移,由于单顺式异构体的最大吸收波长通常有4~6nm的蓝移,因此推断该化合物为单顺式叶黄素;其次四个吸收峰与已知的332nm,418nm,440nm,466nm高度一致;另外q值测定值为0.09与报道的0.10高度一致;最后结合洗脱顺序,综合判断峰9为化合物9-顺式叶黄素,结构见h。
峰10:首先吸收峰的测定值为330nm,420nm,442nm,466nm,其中最大吸收峰442nm于与反式叶黄素的最大吸收峰444nm相比,发生了2nm的蓝移,与已知的单顺式异构体的最大吸收波长通常有4~6nm的蓝移基本一致,推断该化合物为单顺式叶黄素;其次四个吸收峰与已知的332nm,420nm,444nm,472nm及已知的330nm,422nm,440nm,468nm高度一致;另外q值测定值0.09与报道的0.05及报道的0.11高度一致;最后,结合洗脱顺序,综合判断峰10为化合物9’-顺式叶黄素,结构见i。
峰11:首先吸收峰测定值为338nm,446nm,474nm,其中最大吸收峰446与反式玉米黄质的最大吸收峰450nm相比,发生了4nm的蓝移,由于单顺式异构体有4~6nm的蓝移,初步判断为单顺式玉米黄质;其次三个吸收峰与报道的337nm,445nm,473nm及报道的338nm,445nm,472nm高度一致;另外q值测定值为0.09,与报道的0.14的及报道的0.10高度一致;最后,结合洗脱顺序,综合判断峰11为9-顺式玉米黄质。
峰12:首先吸收峰测定值为338nm,446nm,472nm,其中最大吸收峰446与反式玉米黄质的最大吸收峰450nm相比,发生了4nm的蓝移,由于单顺式异构体有4~6nm的蓝移,初步判断为单顺式玉米黄质;其次三个吸收峰与报道的337nm,445nm,473nm及报道的338nm,445nm,472nm高度一致;另外q值测定值0.06与报道的0.14的及0.10基本一致;最后,结合洗脱顺序,综合判断峰12为9’-顺式玉米黄质。
峰1:吸收峰测定值为330nm,410nm,430nm,458nm,其中最大吸收峰430nm与反式叶黄素的最大吸收峰444nm相比,发生了14nm的蓝移,由于双顺式异构体有8~12nm的蓝移,结合出峰顺序初步判断为双顺式叶黄素。
峰2:吸收峰测定值为332nm,410nm,432nm,458nm,其中最大吸收峰432nm与反式叶黄素的最大吸收峰444nm相比,发生了12nm的蓝移,由于双顺式异构体有8~12nm的蓝移,结合出峰顺序初步判断为双顺式叶黄素。
峰4:吸收峰测定值为330nm,418nm,440nm,470nm,其中最大吸收峰440nm与反式玉米黄质的最大吸收峰450nm相比,发生了10nm的蓝移,由于双顺式异构体有8~12nm的蓝移,结合出峰顺序初步判断为双顺式玉米黄质。
表6叶黄素及其异构体的鉴定
各化合物结构如下:
实施例4反式叶黄素定量方法学验证
1系统适用性
按照实施例3的方法分别制备叶黄素对照品溶液及供试品溶液。取对照品溶液连续进样5次测定,记录待测成分峰面积,并计算rsd;取供试品溶液连续进样5次,记录待测成分理论塔板数、分离度、对称因子。结果显示,叶黄素的分离度均大于1.8,已达到基线分离,对称因子1.07(基本符合0.95~1.05的要求),理论板数大于80000,确保定量分析的准确,定为不小于5000,峰面积的rsd为0.26%,小于2%,说明系统适用性较好。
2专属性考察
对照品溶液的制备:按照实施例3中叶黄素对照品溶液的制备方法制备。
供试品溶液的制备:按照实施例2中优化的供试品溶液的制备方法制备。
阴性供试品溶液的制备:取阴性样品粉末,参照实施例2供试品溶液的制备方法制备阴性供试品溶液。
取对照品溶液、供试品溶液、阴性供试品溶液,按照实施例1中所优选的色谱条件分别进,果见图32-34。从图可以看出,供试品色谱中呈现与对照品色谱主峰保留时间一致的色谱峰,空白溶剂及阴性供试品色谱图中在待测成分保留时间处基本无杂质峰出现。表明本方法专属性良好。
3线性及范围
取叶黄素对照品适量,精密称定,加入含0.1%bht的无水乙醇制得每1ml含叶黄素96.5739μg的对照品储备液。分别精密量取该储备液0.2ml、0.4ml、0.8ml、1.2ml、1.6ml、2.0ml于10ml棕色容量瓶中,加入0.1%bht的无水乙醇稀释成1.93、3.86、7.73、11.59、15.45、19.31μg/ml的系列浓度的对照品溶液。
分别吸取上述溶液10μl,按照实施例2的色谱条件进样分析,以对照品溶液浓度(μg/ml)为横坐标(x),以峰面积为纵坐标(y)作图,绘制标准曲线,得回归方程为:y=156255x-7493.5(r2=1),结果表明在1.93~19.31μg/ml的线性范围内,线性关系良好。
4检出限和定量限
取对照品溶液(1.93μg/ml)逐级稀释,以10倍基线噪音(s/n=10)所对应的浓度为定量限,以3倍基线噪音(s/n=3)所对应的浓度为检出限,得到本方法反式叶黄素的定量限为0.019μg/ml,检出限为0.004μg/ml。
5精密度试验
取线性中间浓度(11.59μg/ml)对照品溶液及供试品溶液连续进样6次,测得峰面积,计算rsd,结果对照品和供试品峰面积精密度的rsd均为小于2%,保留时间精密度的rsd也均小于2%,表明仪器精密度良好。
6稳定性试验
分别取同一对照品溶液和同一供试品溶液,分别于0h、1h、2h、3h、4h、5h、6h、9h、12h、15h、19h、24h各进样一次,测定峰面积,计算rsd,对照品溶液峰面积的rsd为0.44%,供试品溶液峰面积的rsd为0.52%,均小于2%。表明对照品溶液和供试品溶液在24h内稳定性良好。
7重复性试验
取同一批次样品,按实施例2方法制备6份供试品溶液,测定,计算样品含量及rsd,结果反式叶黄素的平均含量为5.71%,rsd为0.92%,小于2%。表明该方法的重复性良好。
8加样回收率试验
取叶黄素对照品适量,加入0.1%bht的无水乙醇制成每1ml含反式叶黄素0.6048mg的对照溶液。
取本品粉末150mg,精密称定,置于100ml棕色容量瓶中,加入上述对照品1ml(加入对照品中叶黄素的量与供试品相当),然后按供试品溶液制备方法制备,测定反式叶黄素峰面积,计算加样回收率及rsd,结果见表7。反式叶黄素的平均回收率为101.46%,rsd为0.82%,小于2%。表明该方法的准确性较好。
实施例5三批叶黄素微粒中反式叶黄素的定量测定
1对照品溶液的制备
取叶黄素对照品适量,精密称定,加含0.1%bht的无水乙醇制成每1ml含叶黄素100μg的溶液,即得对照品储备液。分别取储备液0.2、0.4、0.8、1.2、1.6、2.0ml于10ml量瓶中,加0.1%bht的无水乙醇稀释成浓度分别为2、4、8、12、16、20μg/ml的系列浓度的对照品溶液。
2供试品溶液的制备
取三批叶黄素微粒(2017120607、2017120608、2018010602)约300mg,精密称定,置于100ml棕色容量瓶中,加水5ml,于室温下超声(功率100w)处理20min,再加入含0.1%bht的无水乙醇至接近刻度,超声(功率100w)处理5min,冷却至室温,用0.1%bht的无水乙醇定容至刻度,再精密量取1ml至10ml棕色容量瓶中,0.1%bht的无水乙醇稀释至刻度,摇匀,离心,即得。
3色谱条件
以ymccarotenoidc30(250mm×4.6mm,5μm)为色谱柱;以甲醇/水(95:5,含0.1%bht)为流动相a,以甲基叔丁基醚为流动相b,梯度洗脱,即0min~30min、0%~50%b,30min~40min、50%b;流速为0.8ml/min,柱温为30℃,检测波长为445nm。理论塔板数按反式叶黄素峰计算应不低于5000。
4测定
分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
5结果
三批叶黄素微粒的测定结果分别为:2017120607批平均含量为6.05%,2017120608批平均含量为5.71%,2018010602批平均含量为5.68%。
实施例6三批美灵片中反式叶黄素的定量测定
1对照品溶液的制备同实施例5中叶黄素对照品溶液的制备。
2供试品溶液的制备
取三批美灵片(批号分别为:20190101、20190102、20190103),除去包衣,研细,分别取粉末约300mg,精密称定,置于100ml棕色容量瓶中,加水5ml,于室温下超声(功率100w)处理20min,再加入含0.1%bht的无水乙醇至接近刻度,超声(功率100w)处理5min,冷却至室温,用含0.1%bht的无水乙醇定容至刻度,摇匀,离心,即得。
3色谱条件
同实施例5中的色谱条件。
4测定
分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
5结果
三批美灵片的测定结果分别为:20190101批平均含量为4.15mg/g,20190102批平均含量为4.17mg/g,20190103批平均含量为4.20mg/g。
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!