一种同时检测便通胶囊中六种成分含量的方法与流程
本发明属于中药检测领域,涉及一种便通胶囊的六种成分含量的方法。
背景技术:
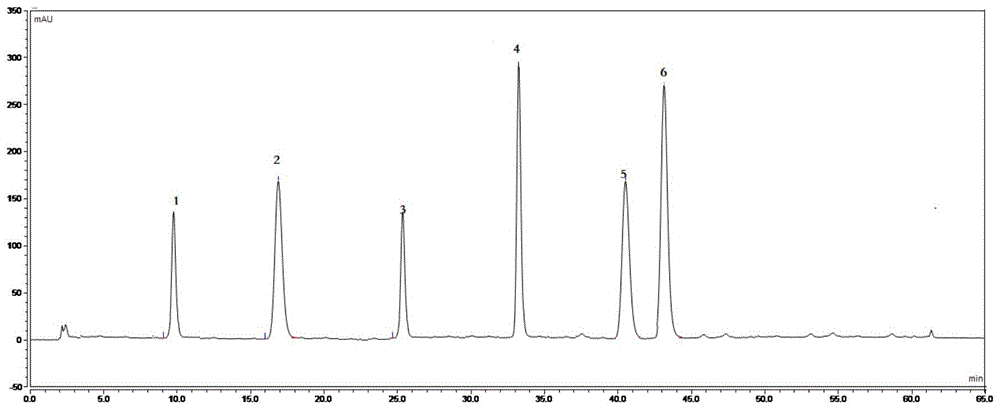
:cn1814131a公开了一种治疗便秘的中药,它由白术、肉苁蓉、当归、桑椹、枳实、芦荟共六味中药材制成,本品为健民集团独家上市品种,商品名“便通胶囊”,主要用于脾肾不足,肠腑气滞所致的便秘。便通胶囊为治疗便秘的常用药品,临床疗效确切,市场需求量大,但其质量标准简单,仅对当归,芦荟,枳实3味药材里的藁本内酯、芦荟苷和辛弗林三种成分进行了薄层定性鉴别,另外还采用hplc法对松果菊苷和芦荟苷进行了含量测定。以上质量控制方法存在以下缺陷:(1)需要对待测品分别进行多次提取和检测,导致检测周期长,需要大量人力物力和检测设备;(2)仅对芦荟苷和松果菊苷进行了定量检测,其它成分均只能定性,从而无法全面控制产品内在质量,不能客观反映和评价中药质量的一致性。因此,迫切需要建立一种针对便通胶囊的新的质量控制方法,以建立可溯源的、与临床疗效高度关联的全程质量控制体系。技术实现要素:本发明的目的是建立一种同时检测便通胶囊中六种成分含量的方法,旨在更高效、可靠地控制产品质量。为实现上述目的,本发明采用如下技术手段:一种同时检测便通胶囊六种成分含量的方法,所述便通胶囊由白术、肉苁蓉、当归、桑椹、枳实、芦荟共六味中药材制成,所述六种成分为阿魏酸、松果菊苷、柚皮苷、毛蕊花糖苷、新橙皮苷、芦荟苷,所述检测方法包括将便通胶囊用提取溶剂提取,然后将提取液上高效液相色谱仪进行检测的步骤,所述高效液相色谱仪含有紫外检测器,所述紫外检测器的设定检测波长280-330nm,所述高效液相色谱仪的固定相为二羟基丙基硅烷键合硅胶色谱柱,流动相a为甲醇,流动相b为0.5-3%(体积)磷酸水溶液,采用梯度洗脱,流速为0.5-2ml/min,柱温为25~35℃。优选地,所述提取溶剂为50%(体积)甲醇。优选地,所述紫外检测器的设定检测波长为280nm和330nm,采用双波长检测,280nm检测阿魏酸,柚皮苷和新橙皮苷三种成分,330nm检测松果菊苷,毛蕊花糖苷和芦荟苷三种成分。优选地,所述流动相b为1%磷酸水溶液。优选地,所述梯度洗脱的时间为65min,分为3个时间段,各时间段流动相a和b的体积变化见下表1:表1梯度洗脱程序时间min流动相a(体积%)流动相b(体积%)0-15257515-4525-5575-4545-6555-9045-10优选地,所述流速为1.0ml/min。优选地,所述柱温为30℃。本发明的有益效果是:(1)本发明能通过一次样品制备和进样同时检测出便通胶囊中六种物质的含量,检测时间不超过2小时,与现有技术相比具有检测耗时少,速度快,费用低,仪器少等优点,大幅提高了检测效率。(2)本发明实现了便通胶囊中六种成分的定量检测,与现有的单一成分定量结合其它成分定性检测相比,本发明能更加全面地控制中药内在质量,客观反映中药质量的一致性,彻底杜绝中药掺假、伪劣现象,更有利于对中药生产的各个阶段包括药材投料等环节进行监控,从而建立可溯源的、与临床疗效高度关联的全程质量控制体系。(3)本发明通过对hplc的检测条件进行大量筛选,使待测六种成分的色谱峰分离效果好、干扰少、出峰时间短、峰面积大,从而提高了检测结果的准确度和灵敏性,使便通胶囊的质量更加可控。附图说明图1是六种混合对照品的hplc图谱。图2是便通胶囊的hplc标准图谱。图中:a是330nm检测波长;b是280nm检测波长。图3是待测样品水超声提取后的hplc图谱。图4是待测样品乙酸乙酯超声提取后的hplc图谱。图5是待测样品50%甲醇超声提取后的hplc图谱。图6是c8色谱柱得到的待测样品hplc图谱。图7是氨丙基色谱柱得到的待测样品hplc图谱。图8是210nm检测波长得到的待测样品hplc图谱。图9是254nm检测波长得到的待测样品hplc图谱。具体实施方式下面通过具体实施例对本发明进行详细地说明。以下实施例中所用到的便通胶囊由健民药业集团股份有限公司生产。处方:麸炒白术296g肉苁蓉210g当归170g桑椹127g枳实127g芦荟65g制法:以上六味,芦荟粉碎成细粉,过筛,备用;其余麸炒白术等六味加水煎煮二次,每次2小时,合并煎液,滤过,滤液静置,取上清液浓缩至清膏状,备用;取上述细粉及淀粉适量,混匀,用上述清膏制粒,颗粒包衣,干燥,装入胶囊,制成1000粒,即得。试药:便通胶囊(批号:180939,由健民药业集团叶开泰国药有限公司提供);芦荟苷对照品(供含测用,中国食品药品检定研究院购买,批号:110787-201808);松果菊苷对照品(供含测用,中国食品药品检定研究院购买,批号:111670-201907);阿魏酸对照品(供含测用,中国食品药品检定研究院购买,批号:110773-201915);柚皮苷(供含测用,中国食品药品检定研究院购买,批号:110722-201815)、新橙皮苷(供含测用,中国食品药品检定研究院购买,批号:111857-201804)、毛蕊花糖苷(供含测用,中国食品药品检定研究院购买,批号:111530-201914)均购自中国食品药品检定研究院;甲醇为色谱纯,磷酸为分析纯,甲醇为分析醇,水为超纯水。实施例1供试品溶液的制备在进行hplc含量测定前,需要对待测药物进行提取,提取的目的一是减少制剂中的检测杂质,包括中药中与检测成分无关的提取成分,以及制剂中的辅料,从而尽可能地减少杂质对待测对象特征峰峰形的干扰,使特征峰具有更好的分离度;二是为了将待测成分尽可能充分地从药物中提取出来,从而提高特征峰的峰面积,保证检测的准确度。我们根据便通胶囊的处方和制剂特点,结合药物成分的溶解性、极性、稳定性,分别考察了三种提取溶剂对检测效果的影响。供试品溶液制备方法:1.取装量差异项下的本品,混匀,研细,取0.5g,精密称定,精密加入水50ml,超声处理(功率250w,频率20khz)30分钟,滤过,取续滤液,即得。2.取装量差异项下的本品,混匀,研细,取0.5g,精密称定,精密加入乙酸乙酯50ml,超声处理(功率250w,频率20khz)30分钟,滤过,取续滤液,即得。3.取装量差异项下的本品,混匀,研细,取0.5g,精密称定,精密加入50%(体积)甲醇50ml,超声处理(功率250w,频率20khz)30分钟,滤过,取续滤液,即得。仪器:安捷伦1260高效液相色谱仪,四元泵,在线真空脱气系统,自动进样器,柱温箱,紫外检测器;ab204-e电子天平(上海梅特勒-托利多仪器有限公司)。流动相:甲醇为流动相a;1%磷酸水溶液为流动相b。梯度洗脱条件:见表1。柱温:30℃;检测波长:330nm;流速:1.0ml/min;进样量:10ul色谱柱:c18色谱柱结果:如图3、4、5所示,1号峰-8min,2号峰-15min,3号峰-24min,4号峰-33min,5号峰-40min,6号峰-43min为有效成分峰,图3(水提取)中4号峰、5号峰、6号峰均未分离。图4(乙酸乙酯提取)中除了4号峰峰面积较大,其他峰峰面积均较小。图5(50%甲醇提取)中有效成分峰均有效分离,峰面积适中,但是峰型待进一步提高。故选择50%甲醇提取。实施例2色谱柱的选择供试品溶液制备方法采用实施例1中的方法3。色谱柱1:c18色谱柱(shim-packveloxc18,5um)色谱柱:2:c8色谱柱(shim-packgistc8,5um)色谱柱3:氨丙基硅烷键合硅胶色谱柱(shim-packgistnh2,5um)色谱柱4:二羟基丙基硅烷键合硅胶色谱柱(shim-packscepterdiol-hilic,5um)其它条件均与实施例1相同。结果:如图2a、5、6、7所示,图5(c18色谱柱)中,1号峰,2号峰,4号峰,5号峰均未达到分离度的要求;图6(c8色谱柱)中,1号峰,3号峰,5号峰,6号峰均未达到分离度的要求;图7(氨基色谱柱)中,峰面积较小,均未达到分离度的要求;图2a(二羟基丙基色谱柱)中,六种成分的峰面积最大,且分离度均大于1.5,故选择二羟基丙基(diol-hilic)色谱柱。表2是四种色谱柱测得的各成分峰分离度。表2四种色谱柱得到的六种成分色谱峰分离度实施例3检测波长的选择供试品溶液制备方法采用实施例1中的方法3。检测波长:210nm、254nm、280nm、330nm固定相:shim-packscepterdiol-hilic其它条件与实施例1相同。结果:从图2、8、9可看出,波长330nm时(图2a)2号峰松果菊苷,4号毛蕊花糖苷,6号芦荟苷峰面积最大,且分离度较好。波长280nm时(图2b)1号峰阿魏酸,3号峰柚皮苷和5号新橙皮苷峰面积最大,且分离度较好。波长210nm时,4号毛蕊花糖苷,5号新橙皮苷,6号芦荟苷均未完全分开。波长254nm时,5号新橙皮苷未完全分离且杂质峰较多。综合考虑,优选双波长检测,280nm检测阿魏酸,柚皮苷和新橙皮苷三种成分,330nm检测松果菊苷,毛蕊花糖苷和芦荟苷三种成分。表3是四种检测波长得到的六种成分色谱峰峰面积。表3四种检测波长得到的六种成分色谱峰峰面积峰面积1-阿魏酸2-松果菊苷3-柚皮苷4-毛蕊花糖苷5-新橙皮苷6-芦荟苷210nm9.0278.89222.038---------254nm8.4259.09511.12667.24756.17666.380280nm17.6978.03624.86167.61773.04651.854330nm8.65322.80511.18387.86138.25486.655实施例4方法验证1.六种物质的定位仪器:安捷伦1260高效液相色谱仪,shim-packscepterdiol-hilic固定相,四元泵,在线真空脱气系统,自动进样器,柱温箱,紫外检测器;ab204-e电子天平(上海梅特勒-托利多仪器有限公司)。流动相:甲醇为流动相a;1%磷酸水溶液为流动相b梯度洗脱条件:见表1。柱温:30℃;检测波长:330nm;流速:1.0ml/min;进样量:10ul对照品溶液的配制:分别精密称取阿魏酸、松果菊苷、柚皮苷、毛蕊花糖苷、新橙皮苷、芦荟苷,置于100ml量瓶中,用甲醇稀释至刻度,配成每1ml含阿魏酸、松果菊苷、柚皮苷、毛蕊花糖苷、新橙皮苷、芦荟苷0.1mg、0.2mg、0.1mg、0.5mg、0.4mg、0.5mg的混合对照品溶液。测定结果见表4,混合对照品的hplc图谱见图1。表4六种物质的定位物质名称对应的峰保留时间相对保留时间分离度阿魏酸1峰9.3750.2841.96松果菊苷2峰16.6350.50373.16柚皮苷3峰25.6420.77642.58毛蕊花糖苷(s)4峰33.02812.86新橙皮苷5峰40.8751.23762.34芦荟苷6峰43.0681.30402.262.线性分别精密称取阿魏酸、松果菊苷、柚皮苷、毛蕊花糖苷、新橙皮苷、芦荟苷对照品各10.0mg,置100ml量瓶中,加甲醇使溶解并定容至刻度,摇匀。分别精密吸取上述对照品溶液0.5,1,1.5,2,2.5,5ml,分别置5ml量瓶中,各加甲醇稀释至刻度,摇匀,各精密吸取10μl,按上述色谱条件测定,以峰积分面积(y:mau*min)对进样量(x:mg)进行线性回归,在定量限浓度至不高于150%指标浓度的范围内取6个浓度点进行研究。线性关系以测得的响应信号(峰面积)对被分析物浓度的函数作图,用最小二乘法进行线性回归,由相关系数r2证实良好的线性关系,要求该线性回归系数r2的数值应不小于0.990,结果见表5。表5六种物质的标准曲线、相关系数及校正因子待测成分标准曲线相关系数校正因子阿魏酸y=894.55x+0.9726r2=0.99960.87松果菊苷y=688.24x-0.1172r2=0.99980.94柚皮苷y=857.91x-0.255r2=0.99760.91毛蕊花糖苷(s)y=2341x-0.5478r2=0.99841.00新橙皮苷y=1385.7x–1.0152r2=0.99740.97芦荟苷y=1875.61x-0.165r2=0.99870.943.精密度①重复性配制6个相同浓度的便通胶囊样品(批号:180939)溶液并进行测试,每个溶液进样2针,要求6次含量测定结果的相对标准差应不大于2.0%,结果见表6。表6六种物质含量测定的重复性试验结果样品阿魏酸松果菊苷柚皮苷毛蕊花糖苷新橙皮苷芦荟苷rsd1.62%1.21%1.02%0.73%0.65%0.86%结果表明:各成分的rsd值在要求范围内,表明检测方法的重复性结果良好。②中间精密度在同一实验室,由不同检验人员于不同日期选择不同仪器对三批样品进行含量测定方法操作,要求两者含量测定结果的相对标准差应不大于2.0%,结果见表7。表7中间精密度考察结果结果表明:各成分的rsd值在要求范围内,方法中间精密度良好4.准确度取已知含量的便通胶囊(批号:180939,已知量按重复性结果计算),研细,分别取约0.25g,0.5g,0.75g,精密称定,置50ml量瓶中,精密量取各对照品溶液25ml,加50%甲醇约20ml,超声处理(功率300w,频率33khz)30分钟,取出,放冷,加50%甲醇至刻度,摇匀,滤过,取续滤液,即得准确度测定用样品溶液;按含量测定方法进行测定,计算准确度,见表8。表8六种物质的回收率测定结果待测成分阿魏酸松果菊苷柚皮苷毛蕊花糖苷新橙皮苷芦荟苷平均回收率97.32%99.26%98.87%98.26%97.86%99.22%结果表明:该方法测定各有关物质的准确度良好。5.稳定性将便通胶囊供试样品(批号:180939)溶液室温保存,每隔4小时或更长时间测定一次峰面积,要求间隔时间内含量测定结果的相对标准差应不大于2.0%,见表9。表9稳定性测定结果结果表明:供试品溶液在24小时内保持稳定。实施例6样品测定(1)供试品溶液的制备:取装量差异项下的本品,混匀,研细,取0.5g,精密称定,精密加入50%甲醇,超声处理(功率250w,频率20khz)30分钟,滤过,取续滤液,即得。(2)对照品溶液的制备:分别精密称取阿魏酸、松果菊苷、柚皮苷、毛蕊花糖苷、新橙皮苷、芦荟苷,置于100ml量瓶中,用甲醇稀释至刻度,配成每1ml分别阿魏酸、松果菊苷、柚皮苷、毛蕊花糖苷、新橙皮苷、芦荟苷0.1mg、0.2mg、0.1mg、0.5mg、0.4mg、0.5mg的混合对照品溶液。(3)色谱条件与系统适应性:仪器:1260高效液相色谱仪,四元泵,在线真空脱气系统,自动进样器,柱温箱,紫外检测器;固定相:shim-packscepterdiol-hilic流动相:甲醇为流动相a;1%磷酸溶液为流动相b梯度洗脱条件:见表1。柱温:30℃;检测波长:280nm,330nm;流速:1.0ml/min;进样量:10ul(4)测定:分别精密吸取对照品溶液与供试品溶液各10μl注入液相色谱仪,测定。(5)计算公式c对为对照品溶液的浓度(mg/ml)a对为对照品溶液的峰面积a样为样品峰面积w样为样品称样量(g)(6)结果:所测得的各有关成分的含量见表10。表105批样品中六种成分的含量(mg/g)批次阿魏酸松果菊苷柚皮苷毛蕊花糖苷新橙皮苷芦荟苷1809340.0240.3820.1221.0120.9871.3621809350.0260.4020.1171.2560.9261.4221809360.0270.3960.1321.0790.9611.4381809370.0230.3930.1261.1530.9791.4261809380.0260.3740.1081.0840.9541.504当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1