一种高效液相色谱法测定华盖散配方颗粒中甘草酸含量的方法与流程
本发明涉及中药成分分析技术领域,具体涉及一种高效液相色谱法测定华盖散配方颗粒中甘草酸含量的方法。
背景技术:
华盖散出自中药经典名方《太平惠民和剂局方》。全方由麻黄,紫苏,杏仁,橘红,桑白皮,茯苓,甘草组成,具有宣肺化痰,止咳平喘的功效,是临床上治疗慢性阻塞性肺病疗效确切的中药组方。
中药配方颗粒是近几年发展起来的利用现代生产技术,以中药饮片为原料,加入适宜的辅料,按照一定生产工艺制成,供临床调剂用的新型颗粒状制剂。
中药复方的配伍使原有某些成分发生量的变化或产生新的化合物,从而表现出增效、减毒、或新的药理活性。在中药煎煮过程中,指标成分产生中和、络合、共溶等化学反应,导致含量与比例的变化,进而造成药理功效的改变。因此,配方颗粒有“单味饮片制剂、临证组方”的特点,决定其缺少水煎剂的合煎过程,进而导致指标成分含量、质量评价方法上存在较大差异。另外,配方颗粒在生产的过程中,存在提取、浓缩、干燥、制粒等过程,样品前处理方法也不同于一般的水煎剂。
由于与传统中药水煎工艺差距较大,有效成分含量可能存在较大的差异,华盖散配方颗粒缺少系统规范的有效成分含量测定方法,导致其质量控制缺乏强有力的理论与科研数据支撑,使用范围受到一定的限制,因此,急需建立一种测定华盖散配方颗粒有效成分的方法。
技术实现要素:
本发明所要解决的技术问题是克服背景技术的技术缺陷,提供一种高效液相色谱法测定华盖散配方颗粒中甘草酸含量的方法。本发明旨在建立一种高效液相色谱法测定华盖散配方颗粒中甘草酸含量的方法,为华盖散配方颗粒质量评价提供更好的理论基础;本发明操作简单,结果可靠、灵敏,满足产品快速检测的需要。
本发明解决上述技术问题所采用的技术方案如下:
一种高效液相色谱法测定华盖散配方颗粒中甘草酸含量的方法,包括样品提取和液相分离两个步骤,对华盖散配方颗粒中的有效成分甘草酸的充分提取和化合物的良好分离,采用美国安捷伦公司的c18反相高效液相色谱柱分离,以甲醇-0.15%甲酸溶液为流动相作梯度洗脱(表1)。
一种高效液相色谱法测定华盖散配方颗粒中甘草酸含量的方法,括如下步骤:
(1)标准溶液的配置:
分别称取甘草酸的对照品适量,精密称定,加甲醇制成含有甘草酸0.4316mg/ml对照品溶液作为储备液,密封,避光保存在4℃冷柜中;
(2)供试品溶液的制备:
取干燥的华盖散配方颗粒粉末0.3g,精密称定,置10ml棕色容量瓶中,加入10ml甲醇,室温浸泡2h后超声30min,重复3次,合并提取液并过滤,回收溶剂后用甲醇溶解并转移至25ml茶色容量瓶,定容并摇匀,经微孔滤膜(0.45μm)滤过,hplc分析;
(3)液相色谱分离:
分离色谱柱:美国安捷伦公司c18反相高效液相色谱柱(5μm,250mm×4.6mm,thermo);
流动相:甲醇(a)-0.15%甲酸(b),梯度洗脱,见表1;
流速:1.0ml/min;
检测波长:250nm;
柱温:30℃;
进样体积:10μl;
表1流动相梯度洗脱程序
(4)含量计算方法:
色谱中甘草酸通过与对照品的液相色谱图和保留时间相比较来确认,按标准曲线法以峰面积计算华盖散配方颗粒中甘草酸的含量。
与现有技术相比,本发明的有益效果是:
(1)本发明样品提取前处理方法简单,有效成分可被充分提取;
(2)本发明准确、快速、灵敏度高,各项方法学指标均可满足实际检测的需要,节约时间与成本;
(3)本发明被测化合物在标准曲线线性范围内呈良好的线性(r2>0.9990);有效成分日内精密度小于1.0%,日间精密度的相对标准偏差均小于3.0%;本发明甘草酸有效成分的回收率在90.0%~110.9%范围内。
附图说明
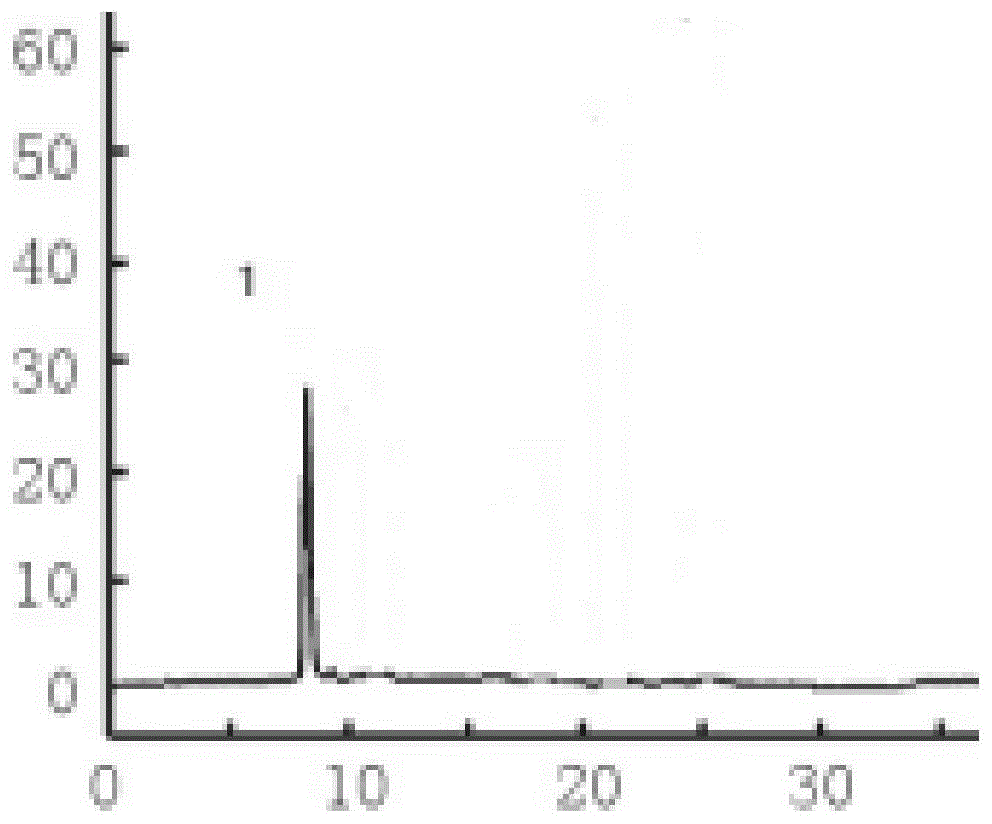
图1为本发明华盖散配方颗粒中甘草酸对照品色谱图;
图2为本发明华盖散配方颗粒中甘草酸供试品色谱图;
图3为本发明华盖散配方颗粒中甘草酸对照品标准曲线图;
图4为本发明华盖散配方颗粒中甘草酸供试品标准曲线图;
其中,图1和图2中的标识为:1-甘草酸。
具体实施方式
为了更好地理解本发明的内容,下面结合具体实施例和附图作进一步说明。应理解,这些实施例仅用于对本发明进一步说明,而不用于限制本发明的范围。此外应理解,在阅读了本发明所述的内容后,该领域的技术人员对本发明作出一些非本质的改动或调整,仍属于本发明的保护范围。
一、实验部分
1.仪器与试剂
高效液相色谱仪:岛津lc-20at高效液相色谱仪(日本岛津公司);岛津pda二级管阵列检测器(日本岛津公司);岛津色谱工作站(日本岛津公司);mettlerab135-s(梅特勒-托利多仪器(上海)有限公司);甲醇为色谱纯(赛默飞化学试剂);其余试剂为分析纯(北京化学试剂厂);水为双蒸水由millipore纯水系统制备(millipore,milford,ma,usa);甘草酸(批号:172146)对照品由中国药品生物制品检验检定研究院提供;以上对照品经hplc分析其纯度均大于98%。
2.液相色谱条件
色谱柱:c18色谱柱(5μm,250mm×4.6mm,安捷伦);
流动相:甲醇(a)-0.05%磷酸水溶液(b),梯度洗脱,所述梯度洗脱程序见表1;
流速:1.0ml/min;
检测波长:250nm;
柱温:30℃;
进样体积:10μl;
表1流动相梯度洗脱程序
(1)标准溶液的配置
称取对照品适量,精密称定,加甲醇制成含有甘草酸0.4316mg/ml对照品溶液作为储备液密封,避光保存4℃冷柜中。
(2)供试品溶液的制备
取干燥的华盖散配方颗粒粉末0.2g,精密称定,置10ml棕色容量瓶中,加入10ml甲醇,室温浸泡2h后超声30min,重复3次,合并提取液并过滤,回收溶剂后用甲醇溶解并转移至25ml茶色容量瓶,定容并摇匀,经微孔滤膜(0.45μm)滤过,hplc分析。
二、结果与讨论
1.提取方法的选择
为了最大程度的提取有效成分甘草酸,试验中比较了提取溶剂、提取方法、提取次数对提取率的影响。首先比较了不同提取溶剂50%甲醇,80%甲醇和纯甲醇对提取率的影响,结果显示纯甲醇提取效率高,基线平,重现好,故选择甲醇作为提取溶剂;随后比较了不同提取方法:超声30min、浸泡1h后超声30min、浸泡2h后超声30min对提取率的影响,结果显示提取效率无显著差异,因此随后的试验都采用超声30min。比较了不同提取次数1次、2次、3次、4次对提取率的影响,结果表明提取3次即可将指标成分完全提取。
2.色谱条件的优化
为了得到一个理想的色谱条件,即在尽量短的时间内使得有效成分的色谱峰与相邻峰之间达到基线分离,试验中对流动相(甲醇-水,乙腈-水,甲醇-乙酸-水,甲醇-甲酸-水,甲醇-磷酸-水)、柱温(25℃,30℃,35℃)等主要影响因素进行了考察。结果表明在上述“2.液相色谱条件”的条件下,甘草酸成分的分离效果最佳。华盖散配方颗粒中指标成分复杂,有高极性化合物,定量成分极性范围大,需要梯度洗脱,且开始梯度变化速率要缓慢,后期需要迅速变化。摸索了多种流动相梯度后,选定的梯度洗脱条件见表1,进行线性回归,分别得到回归方程为:甘草酸:a=652413c+0.3325,在0.0422~0.1436μg范围内峰面积与进样量呈良好的线性关系。
3.标准曲线、检测限和定量限
将对照品储备液逐步稀释得到一系列对照品溶液,以10μl进样测定峰面积值,制备标准曲线,通过峰面积和对照品量作线性回归,计算线性回归和相关系数,结果显示(表2),甘草酸化合物在标准曲线线性范围内呈良好的线性(r2>0.9990)。最低检测限是信噪比为3时样品的量,将信噪比为10时样品的量作为定量限。结果见表2。
表2线性与范围表
4.精密度、重复性和稳定性试验
精密度的考察为测定对照品溶液的日内、日间精密度。日内精密度是在同一天内对同一对照品溶液连续测定6次,而日间精密度则是对同一对照品溶液每天测定一次,连续3天。结果,日内精密度和日间精密度的相对标准偏差分别为0.15%~0.90%和0.98%~2.39%,说明本实验建立的方法具有良好的重现性(表3)。
取同一华盖散配方颗粒6份,按上述方法平行操作、分析、计算,以相对标准偏差来评价该方法的重复性。结果显示(表3),甘草酸化合物的相对标准偏差在1.09%~2.86%的范围内,说明建立的方法重复性良好。取华盖散配方颗粒按照上述方法制备样品溶液,分别在0,2,4,6,8,12,16,24h测定有效成分含量,以相对标准偏差来评价稳定性。结果如表3所示,表明样品在24h内具有良好的稳定性。
表3精密度实验结果
5.回收率试验
回收率试验用来进一步评价方法的准确性。同一样品,按上述供试品制备方法制备相同浓度的6份样品(样品取样量减半,以有效成分1∶1比例加入对照品),通过计算测定的理论值和真实值的比率来计算回收率。结果见表4,甘草酸成分的回收率在90.0%~110.9%范围内,表明该方法具有良好的可靠性和准确性。
表4回收率实验结果
上述说明并非对本发明的限制,本发明也并不限于上述举例。本技术领域的普通技术人员在本发明的实质范围内,做出的变化、改型、添加或替换,也应属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!