同时检测吡喹酮、氯芬新和米尔贝肟三组分含量的方法与流程
1.本发明属于化学分析技术领域,具体涉及一种同时检测吡喹酮、氯芬新和米尔贝肟三组分含量的方法。
背景技术:
2.吡喹酮是一种具有驱虫活性的吡嗪诺喹啉衍生物,在20世纪70年代由拜耳公司研发,是一种广谱抗吸虫和绦虫药。
[0003][0004]
吡喹酮作用机制未被完全阐明,主流的说法认为吡喹酮可能改变虫体对ca
2+
的渗透性,促使其内流导致虫体活动兴奋、肌肉挛缩,从而不能很好吸附于血管壁;另一方面,虫体皮层的损伤也会使得虫体表面的抗原决定簇暴露,从而被宿主免疫系统识别,吸引免疫细胞对其进行攻击。因此,吡喹酮的杀虫机制可概括为药物对虫体的直接作用以及宿主的免疫效应两部分。
[0005]
氯芬新为苯甲酰脲类杀虫剂,是苯甲酰脲类杀虫剂中的第一大产品。氯芬新的主要市场在动物健康领域,用于防治猫、狗身上的跳蚤。
[0006][0007]
此类杀虫剂为昆虫生长调节剂,通过抑制几丁质合成来阻止昆虫外骨骼的进一步生长。该类产品无内吸性,具有胃毒作用。虽然外皮脱落伴随昆虫的整个生命周期,而苯甲酰脲类杀虫剂对幼虫蜕皮阶段最有效。这类产品速效性差,通常与其他传统杀虫剂复配,以提高产品的速效性,增加产品的持效作用。
[0008]
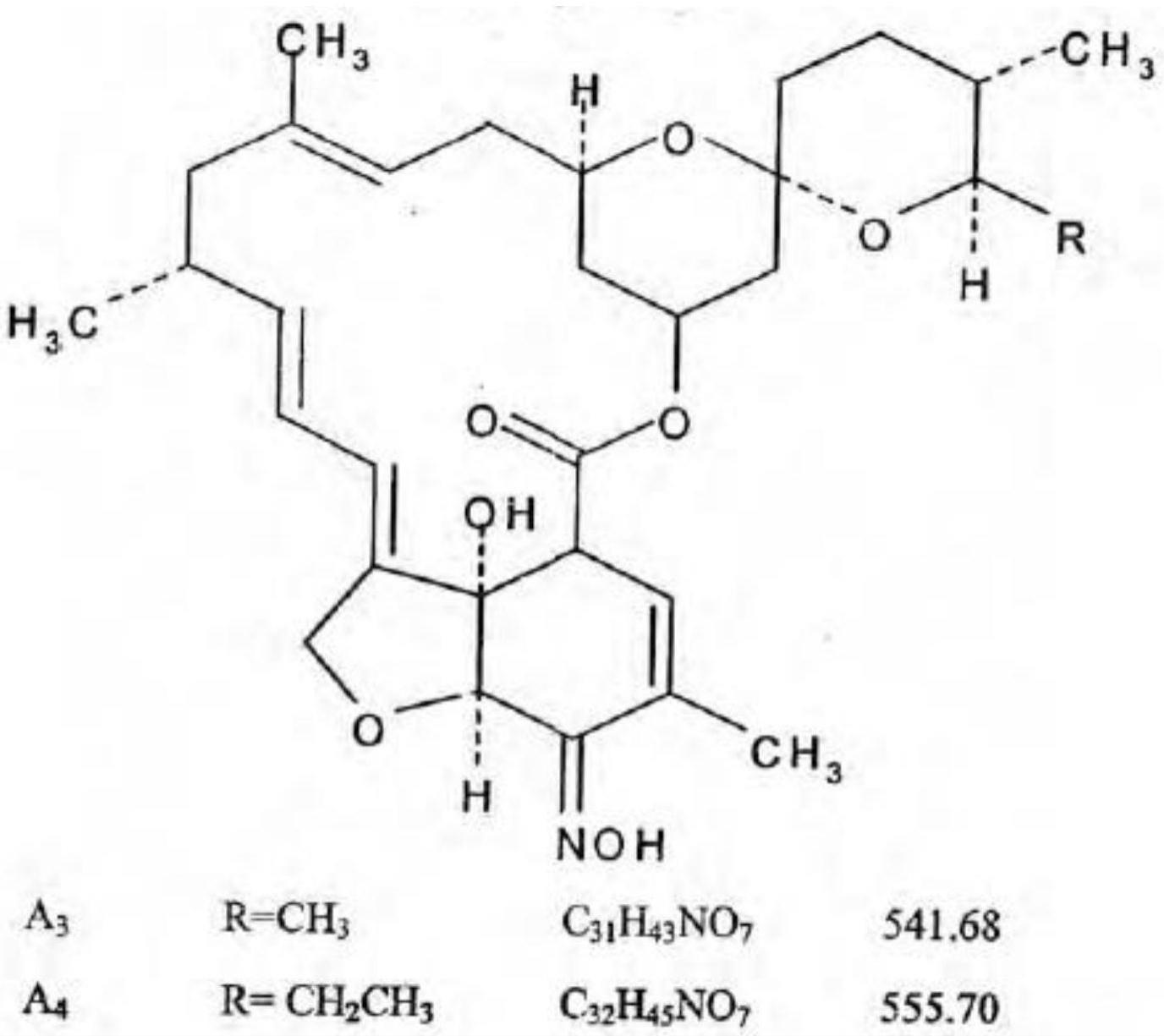
米尔贝肟为半合成大环内酯类抗寄生虫药物,是米尔贝霉素a3和米尔贝霉素a4的肟的衍生物的复合物(比率a4:a3=80:20),由sankyo和ciba-geigy(现在的诺华)共同研发,临床最初用于预防犬心丝虫病和其他体内寄生虫感染的控制。
[0009][0010]
米尔贝肟作用机制与其他大环内酯类抗寄生虫药物类似,米尔贝肟作为神经细胞中gaba(γ-氨基丁酸)神经递质的激动剂,还可与无脊椎动物的神经和肌肉细胞中的谷氨酸门控cl
–
通道结合。在这两种情况下,会阻断寄生虫的神经信号传递,使虫体瘫痪麻痹,导致虫体死亡。米尔贝肟还通过减少虫体产卵或诱导产生异常卵子来影响一些寄生虫的繁殖。但由于绦虫和吸虫虫体内缺少gaba传导介质和谷氨酸门控的cl
–
通道,所以米尔贝肟对绦虫和吸虫无效。
[0011]
以上3个成分杀虫谱互补,联合应用于宠物驱虫有非常良好且广谱的效果。目前海外市场上已出现了以此3个成分配伍的口服宠物驱虫药,但其含量的检测方法并未公开。目前公开的资料中,这三个成分需要用三个不同的hplc方法来检测。
技术实现要素:
[0012]
针对现有技术中的不足,本发明的目的是提供一种同时检测吡喹酮、氯芬新和米尔贝肟三组分含量的方法。
[0013]
为达到上述目的,本发明的解决方案是:
[0014]
一种同时检测吡喹酮、氯芬新和米尔贝肟三组分含量的方法,其包括如下步骤:
[0015]
(1)、量取稀释剂作为空白溶液,注入液相色谱仪中,记录210nm、230nm和255nm波长下的色谱图;
[0016]
(2)、分别称取三组分吡喹酮、氯芬新和米尔贝肟,加稀释剂配制成每1ml含26μg吡喹酮、73μg氯芬新、4.8μg米尔贝肟溶液作为供试品溶液,注入液相色谱仪中,记录210nm、230nm和255nm波长下的色谱图;
[0017]
(3)、取吡喹酮、吡喹酮杂质a、吡喹酮杂质b、吡喹酮杂质c、氯芬新、氯芬新杂质b、氯芬新杂质c、氯芬新杂质g、米尔贝肟及米尔贝肟杂质d的对照品各适量,加稀释剂溶解并
制成每1ml中含上述对照品均为1mg的混合溶液,作为系统适用性试验溶液,注入液相色谱仪中,记录230nm和255nm波长下的色谱图。
[0018]
优选地,步骤(2)中,吡喹酮的浓度为283.20-654.64μg/ml,氯芬新的浓度为570.44-1171.20μg/ml,米尔贝肟的浓度为27.57-64.32μg/ml。
[0019]
优选地,步骤(3)中,吡喹酮杂质a为(11brs)-2-苯甲酰基-1,2,3,6,7,11b-六氢-4h-吡嗪基[2,1-a]异喹啉-4-酮,吡喹酮杂质b为2-(环己甲酰基)-2,3,6,7-四氢-4h-吡嗪并[2,1-a]异喹啉-4-酮,吡喹酮杂质c为n-甲酰基-n-[2-氧-2-(1-氧-3,4-二氢异喹啉-2(1h)-yl)乙基]环己甲酰胺。
[0020]
优选地,步骤(3)中,氯芬新杂质b为n-[(2,5-二氯-4-对羟基苯)氨甲酰基]-2,6-二氟苯甲酰胺,氯芬新杂质c为n-[3-氯-4-(1,1,2,3,3,3-全氟丁二烯丙氧基)苯基-氨甲酰基]-2,6-二氟苯甲酰胺,氯芬新杂质g为2,5-二氯-4-[3-(2,6-二氟苯甲酰)脲基]碳酸二苯酯。
[0021]
优选地,步骤(3)中,米尔贝肟杂质d为(1
′
r,2r,4
′
s,5s,6r,8
′
r,10
′
e,13
′
r,14
′
e,16
′
e,20
′
r,21
′
z,24
′
s)-24
′‑
羟基-21
′‑
(肟基)-6-(丙烷-2-yl)-5,11
′
,13
′
,22
′
四甲基-3,4,5,6-四氢螺环[吡喃-2,6
′‑
[3,7,19]三氧四环[15.6.1.14,8.020,24]二十五烷[10,14,16,22]四烯]-2
′‑
酮。
[0022]
优选地,步骤(1)、步骤(2)和步骤(3)中,稀释剂为乙腈。
[0023]
优选地,步骤(1)、步骤(2)和步骤(3)中,液相色谱仪中用十八烷基硅烷键合硅胶为填充剂nucleosil c18色谱柱(4.6
×
250mm,5μm)。
[0024]
优选地,步骤(1)、步骤(2)和步骤(3)中,液相色谱仪中,梯度洗脱,流速为1.0ml/min,柱温为30℃,紫外检测波长为230nm,进样量20μl;流动相a为0.01%(v/v)磷酸溶液,流动相b为乙腈,流动相a的含量为10-30wt%,流动相b的含量为70-90wt%。
[0025]
由于采用上述方案,本发明的有益效果是:
[0026]
本发明的检测方法简便,节约时间,降低成本,有很好的专属性、准确度、线性、耐用性及精密度,用于检测三组分的含量是适宜的、准确的,从而可用于宠物药日常检测的需要。
附图说明
[0027]
图1为本发明的210nm的空白溶液色谱图。
[0028]
图2为本发明的230nm的空白溶液色谱图。
[0029]
图3为本发明的255nm的空白溶液色谱图。
[0030]
图4为本发明的210nm的供试品溶液色谱图。
[0031]
图5为本发明的230nm的供试品溶液色谱图。
[0032]
图6为本发明的255nm的供试品溶液色谱图。
[0033]
图7为本发明的230nm的系统适用性试验溶液色谱图。
[0034]
图8为本发明的255nm的系统适用性试验溶液色谱图。
[0035]
图9为本发明的实施例3中吡喹酮平均峰面积对浓度曲线图。
[0036]
图10为本发明的实施例3中氯芬新平均峰面积对浓度曲线图。
[0037]
图11为本发明的实施例3中米尔贝肟平均峰面积对浓度曲线图。
具体实施方式
[0038]
本发明提供了一种同时检测吡喹酮、氯芬新和米尔贝肟三组分含量的方法。
[0039]
根据文献可知:吡喹酮(praziquantel)紫外检测波长为210nm
[1]
、氯芬新(lufenuron)紫外检测波长为255nm
[2]
、米尔贝肟(milbemycin oxime)紫外检测波长为230nm
[3]
。
[0040]
本发明的同时检测吡喹酮、氯芬新和米尔贝肟三组分含量的方法包括如下步骤:
[0041]
(1)、稀释剂乙腈为空白溶液,精密量取20μl,注入液相色谱仪,记录3个紫外波长(210nm、230nm和255nm)下的色谱图,如图1至图3所示。
[0042]
(2)、分别称取三组分,加乙腈配制成每1ml含吡喹酮约26μg、氯芬新约73μg、米尔贝肟约4.8μg溶液作为供试品溶液,精密量取20μl,注入液相色谱仪,记录3个紫外波长(210nm、230nm和255nm)下的色谱图,如图4至图6、表1至表3所示。
[0043]
(3)、取吡喹酮、吡喹酮杂质a、吡喹酮杂质b、吡喹酮杂质c、氯芬新、氯芬新杂质b、氯芬新杂质c、氯芬新杂质g、米尔贝肟及米尔贝肟杂质d的对照品各适量,加乙腈溶解并稀释制成每1ml中约含上述对照品均为1mg的混合溶液,作为系统适用性试验溶液。精密量取20μl,注入液相色谱仪,记录紫外波长230nm和255nm下的色谱图,如图7和图8、表4和表5所示。
[0044]
由图1至图3可知,空白溶液-乙腈峰不干扰各组分峰(组分峰都在5分钟之后出峰)。
[0045]
由图4至图6可知,210nm的吡喹酮与氯芬新峰面积相对最大,但210nm对于米尔贝肟是末端吸收、峰面积小,因而不选用;吡喹酮在255nm的峰面积最小,其他组分在230nm与255nm的峰面积相差不大,所以选择230nm为检测波长。
[0046]
由图7至图8可知,两波长检测结果相比较可知,在230nm波长下,系统适用性试验溶液中各组分峰与前后杂质峰之间的分离度均大于2.0,且各组分峰的理论板数均大于5000。因此采用230nm为三组分含量测定的检测波长。
[0047]
表1
[0048]
description dad1a,sig=210,4ref=off
[0049][0050]
表2
[0051]
description dad1b,sig=230,4ref=off
[0052]
[0053]
表3
[0054]
description dad1c,sig=255,4ref=off
[0055][0056]
表4
[0057]
description dad1b,sig=230,4ref=off
[0058][0059][0060]
表5
[0061]
description dad1c,sig=255,4ref=off
[0062][0063]
该色谱条件下,四个主成分峰-吡喹酮、氯芬新、米尔贝肟a3、米尔贝肟a4能完全分离,各杂质对四个主成分峰无干扰,重复性试验吡喹酮rsd为0.38%(n=6)、氯芬新rsd为0.42%(n=6)、米尔贝肟rsd为0.26%(n=6)。
[0064]
其中,
[0065]
1)根据欧洲药典ep9.0
[1]
可知,吡喹酮的已知杂质有:吡喹酮杂质a、吡喹酮杂质b、吡喹酮杂质c。
[0066]
吡喹酮杂质a:(11brs)-2-苯甲酰基-1,2,3,6,7,11b-六氢-4h-吡嗪基[2,1-a]异喹啉-4-酮,是生产过程中产生的合成杂质。
[0067][0068]
吡喹酮杂质b:2-(环己甲酰基)-2,3,6,7-四氢-4h-吡嗪并[2,1-a]异喹啉-4-酮,是生产过程中产生的合成杂质。
[0069][0070]
吡喹酮杂质c:n-甲酰基-n-[2-氧-2-(1-氧-3,4-二氢异喹啉-2(1h)-yl)乙基]环己甲酰胺,是吡喹酮的降解杂质。
[0071][0072]
2)根据美国药典usp43
[2]
可知,氯芬新的已知杂质有:氯芬新杂质b、氯芬新杂质c、氯芬新杂质g。
[0073]
氯芬新杂质b:n-[(2,5-二氯-4-对羟基苯)氨甲酰基]-2,6-二氟苯甲酰胺,是生产过程中产生的合成杂质。
[0074][0075]
氯芬新杂质c:n-[3-氯-4-(1,1,2,3,3,3-全氟丁二烯丙氧基)苯基-氨甲酰基]-2,6-二氟苯甲酰胺,是生产过程中产生的合成杂质。
[0076][0077]
氯芬新杂质g:2,5-二氯-4-[3-(2,6-二氟苯甲酰)脲基]碳酸二苯酯,是生产过程中产生的合成杂质。
[0078][0079]
3)根据文献
[3]
可知,米尔贝肟的已知杂质有:米尔贝肟杂质d。
[0080]
米尔贝肟杂质d:(1
′
r,2r,4
′
s,5s,6r,8
′
r,10
′
e,13
′
r,14
′
e,16
′
e,20
′
r,21
′
z,24
′
s)-24
′‑
羟基-21
′‑
(肟基)-6-(丙烷-2-yl)-5,11
′
,13
′
,22
′
四甲基-3,4,5,6-四氢螺环[吡喃-2,6
′‑
[3,7,19]三氧四环
[0081]
[15.6.1.14,8.020,24]二十五烷[10,14,16,22]四烯]-2
′‑
酮(milbemycin d oxime),是生产过程中产生的合成杂质。
[0082][0083]
具体验证过程如下:
[0084]
检测方法描述
[0085]
色谱柱:用十八烷基硅烷键合硅胶为填充剂(nucleosil c18 250
×
4.6mm,5μm或效能相当的色谱柱)。
[0086]
流速:1.0ml/min。
[0087]
柱温:30℃。
[0088]
波长:230nm。
[0089]
进样体积:20μl。
[0090]
流动相:以0.01%(v/v)磷酸溶液为流动相a,乙腈为流动相b,按表6进行线性梯度洗脱。
[0091]
表6
[0092][0093]
稀释剂:乙腈
[0094]
出峰顺序:吡喹酮、氯芬新、米尔贝肟a3、米尔贝肟a4。
[0095]
米尔贝肟对照品储备液:精密称定米尔贝肟对照品(来源:usp,批号:r086q0,含量:97.4%)23.0mg,置10ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,即得。
[0096]
对照品溶液(临用新制):精密称定吡喹酮对照品(来源:中国药品生物制品检定所,批号:100046-201205,含量:99.7%)23mg、氯芬新对照品(来源:国家农药质量监督检验中心(沈阳),批号:20190601,含量:98.4%)46mg,再精密量取米尔贝肟对照品储备液1.0ml,置50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,即得。
[0097]
供试品溶液(临用新制):浓度与对照品溶液的一致。
[0098]
以下结合实施例对本发明作进一步的说明。
[0099]
实施例1:系统适用性
[0100]
色谱分析程序
[0101]
按照分析方法的要求做系统适用性试验。在系统平衡后,取对照品溶液20μl,注入液相色谱仪,记录的色谱图。先进5针std-1,计算相对标准偏差(rsd),再进2针std-2,用std-1计算std-2的准确度,如表7。
[0102]
表7
[0103][0104][0105]
用std-1计算std-2的回收率的公式:
[0106][0107]astd-1
=std-1色图谱中各组分峰面积的平均值。
[0108]astd-2
=std-2色图谱中各组分峰面积的平均值。
[0109]cstd-1
=std-1中各组分的浓度。
[0110]cstd-2
=std-2中各组分的浓度。
[0111]
系统适用性试验结果,如表8至表11。
[0112]
表8
[0113][0114]
表9
[0115][0116]
表10
[0117][0118][0119]
表11
[0120][0121]
综上可知:吡喹酮5针std的rsd为0.056%,远远小于2.0%,std-2对于std-1的回收率为99.4%;氯芬新5针std的rsd为0.10%,远远小于2.0%,std-2对于std-1的回收率为100.3%;米尔贝肟5针std的rsd为0.075%,远远小于2.0%,std-2对于std-1的回收率为100.5%。
[0122]
实施例2:专属性
[0123]
专属性将通过空白的干扰和降解杂质的干扰性来确认。
[0124]
空白溶液(稀释剂):取乙腈直接进样。
[0125]
样品:分别称取吡喹酮原料(来源:河北嘉一药业有限公司,批号:jyc-200402)、氯芬新原料(来源:浙江海晟,批号:n181101)、米尔贝肟原料(来源:浙江海正药业,批号:4037-a180405u)组成混合物的相近重量14.2mg作为破坏试验样品。
[0126]
酸降解:称取样品,置100ml量瓶中,加5.0ml、1mol/l的盐酸溶液,于室温下放置5min后,用1mol/l的氢氧化钠溶液调节ph值至7.0,冷却至室温,用稀释剂稀释至刻度,摇匀,即得。
[0127]
碱降解:称取样品,置100ml量瓶中,加5.0ml、1mol/l的氢氧化钠溶液,于室温下放置5min后,用1mol/l的盐酸溶液调节ph值至7.0,冷却至室温,用稀释剂稀释至刻度,摇匀,即得。
[0128]
氧化降解:称取样品,置100ml量瓶中,加5.0ml、30%的过氧化氢溶液,于室温下立即振摇后,用稀释剂稀释至刻度,摇匀,即得。
[0129]
热降解:将样品置于透明玻璃瓶中密封,暴露在90℃的高温条件下24h,冷却至室温后,按照供试品溶液配制方法配制样品溶液。
[0130]
高湿降解:将样品置于透明玻璃瓶中密封,暴露在92.5%rh的高湿条件下10天后,取出,按照供试品溶液配制方法配制样品溶液。
[0131]
专属性研究试验结果如表12。
[0132]
表12
[0133][0134]
综上可知:空白溶液在各主峰的位置不出峰,不存在干扰;在所有强力破坏试验下,被检测出的降解峰能与各主峰完全分离,各主峰与前后相邻杂峰的分离度均大于2.0;米尔贝肟酸降解、碱降解条件降解达30%以上,但系统适用性良好且不存在干扰,说明此方法专属性良好。
[0135]
实施例3:线性
[0136]
在含量测定中吡喹酮、氯芬新和米尔贝肟的线性研究的测定浓度范围为60-140%。按如下要求进行线性研究试验。
[0137]
测试程序和结果处理
[0138]
以浓度(μg/ml)为横坐标、峰面积为纵坐标,绘制标准曲线y=ax+b。
[0139]
计算标准曲线的相关系数r。
[0140]
计算线性偏离bias%:即截距b相对于100%浓度时峰面积y
100%
的百分率。
[0141][0142]
计算平均响应因子(mrf):
[0143][0144]
线性试验结果如表13至表15、图9至图11所示。
[0145]
表13
[0146]
[0147][0148]
表14
[0149]
[0150][0151]
表15
[0152][0153]
综上可知:各浓度下重复三次进样的重复性rsd
n=3
都小于2.0%,说明在各浓度下该方法都有很好的系统精密度;相关系数r:为0.9997(吡喹酮)、0.9991(氯芬新)和1(米尔贝肟),均大于0.999,说明方法有很好的线性回归;线性偏离bias%:为1.7%(吡喹酮)、1.8%(氯芬新)和0.00041%(米尔贝肟),均小于2.0%,说明本方法的系统误差较小。
[0154]
通过以上可知,吡喹酮在283.20-654.64μg/ml浓度下有很好的线性和系统精密
度;氯芬新在570.44-1171.20μg/ml浓度下有很好的线性和系统精密度;米尔贝肟在27.57-64.32μg/ml浓度下有很好的线性和系统精密度。
[0155]
实施例4:准确度
[0156]
在空白溶液中加入相当于自身对照浓度60%、100%、140%的三组分后,通过回收率来评价本方法准确度。
[0157]
米尔贝肟对照品储备液:精密称定米尔贝肟对照品23.0mg,置10ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,即得。
[0158]
对照品溶液:精密称定吡喹酮对照品23mg、氯芬新对照品46mg,再精密量取米尔贝肟标准品储备液1.0ml,置50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,即得。
[0159]
自身对照储备液:取吡喹酮对照品230mg、氯芬新对照品460mg,精密称定,置50ml的量瓶中,加稀释剂溶解并稀释至刻度,摇匀,即得。
[0160]
60%加标样品溶液:精密量取自身对照储备溶液6.0ml与米尔贝肟对照品储备液1.2ml,置100ml量瓶中,加稀释液稀释至刻度,摇匀。共3份。
[0161]
100%加标样品溶液:精密量取自身对照储备溶液10.0ml与米尔贝肟对照品储备液2.0ml,置100ml量瓶中,加稀释液稀释至刻度,摇匀。共3份。
[0162]
140%加标样品溶液:精密量取自身对照储备溶液14.0ml与米尔贝肟对照品储备液2.8ml,置100ml量瓶中,加稀释液稀释至刻度,摇匀。共3份。
[0163]
按对照品溶液,60%加标溶液,100%加标溶液,140%加标溶液,依次进样,对照品溶液重复进样5次,其余溶液重复进样2次,按外标法计算各级加标样品溶液的加标回收率如表16至表18。
[0164]
表16
[0165][0166]
表17
[0167]
[0168][0169]
表18
[0170][0171]
综上可知:60%、100%、140%浓度下的加标回收率:都在98.0-102.0%之间,说明在各浓度下都有很好的回收率;同一浓度下的3次试验回收率的rsd%
n=3
均远远小于2.0%;不同浓度间,平均回收率的rsd%
n=3
均远远小于2.0%。
[0172]
通过以上可知,吡喹酮在283.20-654.64μg/ml浓度下,氯芬新在570.44-1171.20μg/ml浓度下,米尔贝肟在27.57-64.32μg/ml浓度下,即相当于含量的60-140%,有很好的准确度。
[0173]
实施例5:精密度
[0174]
方法重复性
[0175]
平行配制6份样品(相同批次三组分的混合物)溶液,按照检测方法的要求进行三组分含量检测。以评价本方法在相同实验环境下,对测试同一批样品时结果的重复性。
[0176]
方法中间精密度
[0177]
中间精密度所用的6份样品溶液采用和重复性试验相同批号的样品,按要求配制样品溶液。应由不同实验员在不同日期,用不同仪器来进行检测,时间间隔至少24h。以证明本方法在可变实验环境下,对同一批样品测试时结果的重现性。精密度试验结果如表19。
[0178]
表19
[0179][0180]
综上可知:同一天方法重复性试验:分别6个检测结果中,rsd%
n=6
均未超过0.42%,远远小于2.0%,说明本方法有很好的重复性。不同天不同测试人员重复性试验:共12个检测结果中,rsd%
n=12
均未超过0.45%,远远小于2.0%,说明本方法有很好的方法中间精密度。
[0181]
通过以上可知,本方法有很好的重复性和精密度。
[0182]
实施例6:耐用性研究
[0183]
分别在改变以下某一色谱参数而其它因素(仪器、人)不变的条件下,对关键参数进行确认,如发现有较大的影响,则该影响因素需要在分析方法设置里进行严格控制。
[0184]
改变柱温(
±
2℃)。
[0185]
改变波长(
±
2nm)。
[0186]
改变流速(
±
0.2ml/min)。
[0187]
改变色谱柱(不同厂家同规格:luna c18(2)250
×
4.6mm,5μm)。
[0188]
色谱分析程序:每种hplc条件按照含量分析方法中所述进行系统适用性试验,然后对同一批样品进行含量的测定。系统适用性耐用性试验结果如表20。
[0189]
表20
[0190]
[0191][0192]
综上可知:每个变化条件下所测的含量结果与初始条件下测得结果差异均在2.0%以内,每个变化条件下std-1峰面积的rsd均在2.0%以内,每个变化条件下用std-1计算std-2的回收率均在98.0-102.0%之间。说明检测方法在细微变化后,仍能保证含量检测结果的一致性,用该方法检测三组分含量,有很好的耐用性。
[0193]
实施例7:含量方法验证
[0194]
本含量检测方法参照欧洲药典ep9.0兽用氯芬新质量
[4]
标准制定。对本发明的含量方法进行了方法学验证
[5]
。
[0195]
研究发现在规定的色谱条件下,主峰与各杂质峰分离良好。验证结果显示本方法有很好的专属性、准确度、线性、耐用性及精密度,用于检测三组分的含量是适宜的、准确的。
[0196]
含量验证的结果总结表见表21。
[0197]
表21含量方法学验证结果表
[0198]
[0199][0200]
由表21可知,该含量分析方法的验证结果显示本方法有很好的专属性、准确度、线性、耐用性及精密度,用于同时检测三组分的含量是适宜的、准确的。
[0201]
参考文献:
[0202]
[1]《欧洲药典》ep9.0版吡喹酮质量标准。
[0203]
[2]《美国药典》usp43版氯芬新质量标准。
[0204]
[3]《农业部第2272号公告》米尔贝肟吡喹酮片质量标准。
[0205]
[4]《欧洲药典》ep9.0版兽用氯芬新质量标准。
[0206]
[5]《中国兽药典》2020年版一部附录9101兽药质量标准分析方法验证指导原则。
[0207]
上述对实施例的描述是为了便于该技术领域的普通技术人员能理解和使用本发明。熟悉本领域技术人员显然可以容易的对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中,而不必经过创造性的劳动。因此,本发明不限于上述实施例。本领域技术人员根据本发明的原理,不脱离本发明的范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1